New horizons in next-generation small molecule kinase inhibitors
Posted: 16 June 2016 | Karolina Gluza (Selvita), Monika Dobrzańska (Selvita) | No comments yet
In the pre-imatinib (Glivec®) era, two out of three patients died within five years from chronic myeloid leukemia and there was no hope for a curative treatment. Imatinib’s introduction in 2001 marked a breakthrough – as the first approved kinase inhibitor, it was a game-changer in cancer therapy and revolutionised the approach to treating malignancies, serving as the first genetically tailored cancer drug. Precision medicine is certainly not a novel concept, but kinase inhibitors have given it a huge boost.
The clinical proof-of-concept delivered by kinase inhibitors (‘the right drug for every patient and the right patient for every drug’) facilitated rapid expansion of the therapeutic arsenal for malignancies, inflammation and autoimmune disorders. 15 years later, there is still excitement in the field with novel perspectives and opportunities on the horizon.
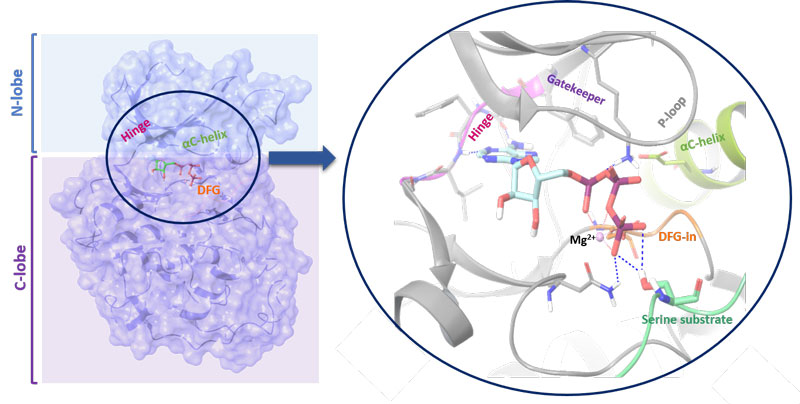
Figure 1: Structure of a kinase. The N-lobe is connected by a hinge to the C-lobe. The position of the C-helix together with the Asp-Phe-Gly (DFG) motif (in-out mode) serves as indicator of active/inactive kinase state. The ATP binding cleft and gatekeeper are also shown
Kinases are molecular on-off switches and orchestrate signalling cascades regulating essential cellular pathways. Functionally, the enzymes transfer a γ-phosphate group from adenosine triphosphate (ATP) or other high-energy donors to the hydroxyl group of its amino acid, sugar or lipid substrates. Kinases are structurally diversified with the primary amino acid sequence they adopt conserved, bilobal architectures which enable catalytic activity (see Figure 1; page 28). To describe their structure in more detail, the smaller, N-terminal lobe (N-lobe) is more flexible and consists mainly of β-strands. It is connected by a hinge with a larger, more static C-terminal lobe (C-lobe) which is primarily α-helical and contains a control helix (C-helix). It possesses conservative glutamic acid that forms a salt bridge with catalytic lysine residue. The position of the C-helix together with the Asp-Phe-Gly (DFG) motif (in-out mode) serves as an indicator of active/inactive kinase state. The ATP binding cleft is located between both lobes and the hinge region contains a gatekeeper. Its size and the chemical character influences the compound binding, due to fact that it controls the access to the back pocket region of the enzyme and often directly interacts with small molecule inhibitor.
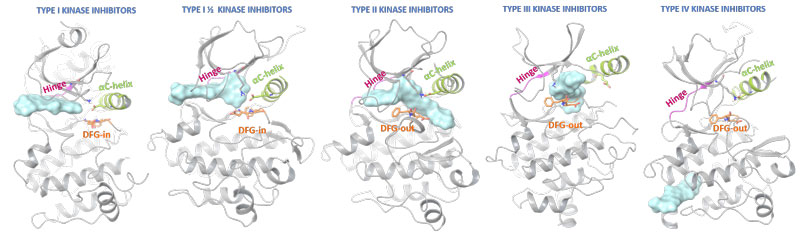
Figure 1: Structure of a kinase. The N-lobe is connected by a hinge to the C-lobe. The position of the C-helix together with the Asp-Phe-Gly (DFG) motif (in-out mode) serves as indicator of active/inactive kinase state. The ATP binding cleft and gatekeeper are also shown
Ligand-protein interactions are not limited to the active site and strongly depend on the kinase state (Figure 2). Most approved protein kinase inhibitors represent type I interactions, competing with ATP for the adenine binding pocket in an activated kinase. Type II inhibitors also occupy similar protein regions, but additionally bind adjacent allosteric pockets created by DFG out-conformations. Type I ½ kinase modulators combine binding characteristic for both type I and II modes, and types III and IV utilise additional protein areas to silence kinase signalling. Type V include bivalent or bisubstrate inhibitors (not shown in diagram) and also cover hybrid type I ½ kinase modulators.
There are 518 kinases in the human genome[i]. The variety of malignancies and significant range of other life-threating and chronic diseases is related to aberrant kinase signalling. In particular, uncontrolled kinase-dependent pathways have been shown to be major drivers of cancer hallmarks like proliferation, survival, motility, metabolism, angiogenesis and attenuation of immune-mediated antitumour responses[ii]. Figure 3 summarises United States Food and Drug Administration-approved small molecule kinase inhibitors.
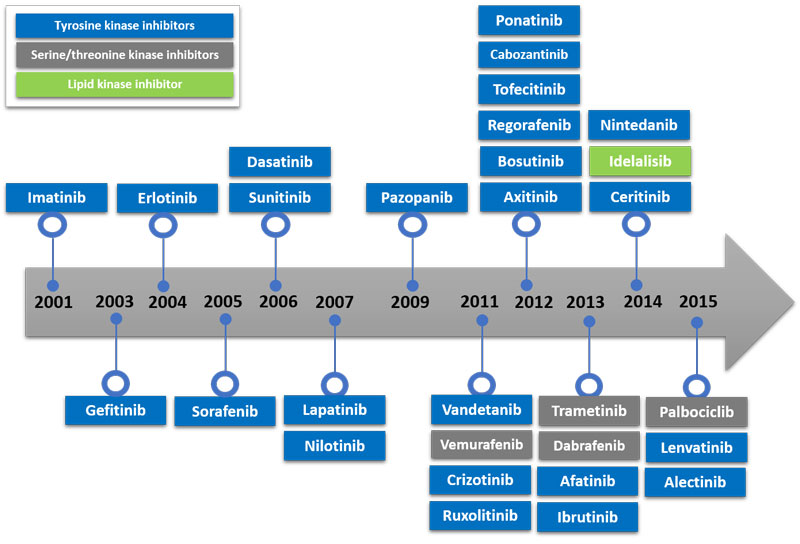
Figure 3: FDA-approved small molecule kinase inhibitors
Limitations of kinase inhibitors
Although kinase inhibitors represent an advancement in cancer therapy, they still do not benefit every patient. Strikingly, the major issue lies in their strength, making the targeted therapy a double-edged sword. The underlying success of a personalised therapy lies in a fact it targets one cancer-specific kinase. However, in a constant battle for supremacy, tumour-dependent kinases acquire recurrent mutations affecting amino acid sequence, conformation, folding or expression level. The small molecule drug cannot access the binding site or compete with ATP and interfere with protein signalling, which ultimately leads to a lack of kinase inhibitors’ efficacy. BRAF V600, KIT D816, FLT3-ITD, FLT3D835 or MET M1268T mutations may serve as clinically relevant resistance examples. It highlights strong cancer dependence on a specific kinase, but on the other hand creates escape mechanisms.
The second limitation tightly accompanies the former one. Kinases may mediate disease-related signalling, but the hunt is on to identify the crucial molecular player with good translation from preclinical efficacy to real life treatment advantages. The cancer landscape is complex, and strong one-kinase addiction is not common, so unfolding the malignancy-kinase link may be challenging[iii]. From the recently approved kinase inhibitors, the only drug that appears able to replicate the imatinib success story is ibrutinib (Imbruvica®). It is clear the field still requires identification of novel, putative driver kinases supported by comprehensive sequencing technologies, genomic analyses searches and identification of biomarkers which can predict therapeutic responsiveness.
Promising pipelines
New possibilities await exploration since only a small fraction of the human kinome has been intensively studied and efforts have been heavily focused on tyrosine kinase (TK) inhibitors. This disproportion is clearly illustrated by the fact that TK inhibitors of BCR-Abl, ErbB and VEGFR account for 18 of almost 30 approved protein kinase inhibitors in total. However, ongoing clinical trials are evaluating a variety of novel targets, some with ‘first in class’ potential, to name but a few: the Aurora subfamily, CDK family, TGFβ kinase, SYK, PLK1, Rho kinases (ROCK) 1 and 2, PDKs, S6 kinase subfamily, CDC7, GSK3, WEE1, PIM1-3, PTK2, MAPKs (MLK, MAP3K9, MAP3K10, MAP3K11) and CHK1 and 2 (reviewed in [iv]).
Additionally, targeting of pseudokinases shows promise. Despite the lack of essential catalytic residues to effectively transfer phosphate, there is evidence they regulate activity of other kinases[v],[vi]. Proteins such as STRAD, HER3, JAK1-3, TYK2, IRAK2, CCK4 and GCN2, which possess pseudokinase subunits, may serve as master regulators of functional kinase signalling and potentially offer superior therapy to standard inhibitors of kinase catalytic activity[vii].
It is not only the kinome that is underexplored. The therapeutic application of current kinase inhibitors is limited to cancer, with the two exceptions of tofacitinib (Xeljanz®), approved for arthritis and nintedanib (Ofef®), which is prescribed for idiopathic pulmonary fibrosis. However, this group of compounds holds great potential for the future.
With new immunotherapies, the adaptive anti-cancer mechanism evolves and accommodates as a patient’s individual cancer evolves and mutates. The first checkpoint inhibitor ipilimumab (Yervoy®; anti-CTLA-4) entered the immunooncology field in 2011 (followed by anti-PD1 pembrolizumab; Keytruda®), ten years after imatinib was approved. These two new approaches revolutionised the way in which malignancies are treated today. Kinase inhibitors give rise to a relatively immediate response, but have an issue with prolonging the survival rate, whereas immunotherapeutics take a while to show clinical benefits, but the response is durable over time. Therefore, immunokinases may be a groundbreaking ‘one size fits all’ solution. They directly target immune cells to enhance cancer recognition, elimination or modulate tumour microenvironment, controlling cytokines and chemokines release.
The kinases involved in T cell receptor signalling and T cell proliferation, such as LCK or ITK[viii], may play a relevant role. DGK is crucial in cancer-related immune surveillance and shapes the immune response in addition to having a direct effect on Ras[ix]. HPK1 negatively regulates dendritic cells (antigen presentation) and T cell (proliferation) functions through AP-1, NFAT and NFκB pathways[x],[xi]. The PI3K–AKT–mTOR axis is directly involved in the induction of anergy in T cells, independent of the cell cycle regulation. mTOR inhibition was also shown to induce differentiation of CD4 cells into CD4+CD25+Foxp3+Treg cells[xii],[xiii]. IPI-549 is a selective PI3K-γ inhibitor which promotes proliferation of CTLs by modulating the immune-suppressive tumour microenvironment[xiv],[xv]. Treg functions and development is also affected by protein kinase C (PKC) and proto-oncogene tyrosine-protein kinase Src (c-SRC kinase; CSK)[xvi].
CSF1R inhibitors repolarise tumour-associated macrophages, resulting in antitumour efficacy with prolonged overall survival as single-agent therapy or in combination with checkpoint inhibitors PD-1 or CTLA4[xvii],[xviii],[xix]. The TGF-β kinase inhibitor galunisertib showed remarkable therapeutic potential in vivo[xx]. Kinase players from BCR, Toll-like receptors (TLR) and interferons signalling pathways may also have a substantial influence on tumour immunity, e.g., BTK, SYK, Lyn, TAK1, IKK kinases, TYK2, JAK1-3 or IRAK4 and 1. The same set of kinases is essential for chronic inflammatory diseases. The examples given constitute only a minor, representative portion of the great immunoncology potential hiding within the unexplored kinome.
Silencing of kinase signalling may be also advantageous in non-life threatening disorders (for review see 4). For example, the ROCK 1 and 2 small molecule inhibitors are emerging treatment options for patients with glaucoma allowing them to prevent or delay optic nerve damage (they are currently in clinical trials)[xxi]. Parkinson’s disease patients may one day benefit from drugs that modulate the kinases JNK1-3, ERKs, MAPKs, LRRK2 and MLKs[xxii],[xxiii],[xxiv]. The landscape of Alzheimer’s disease may also be changed due to GSK3 and PKC[xxv]. Meanwhile, mTOR inhibition brings benefits after percutaneous coronary intervention with additional perspectives for PKC kinases and fibroblast growth factor receptor 4 (FGFR4) in cardiovascular disorders. An intriguing potential in osteoporosis is served by activin receptor type IIB (ActRIIA) kinase[xxvi]. It is clear from these examples that there is a growing interest in kinase inhibition for chronic disorders.
In the early days, robust expansion of clinical kinase inhibitors resulted in fine-tuned properties and more therapeutics readily available for patients, but for the price of modest structural diversity (gefinitinib- and imatinib-like). The trend of the ‘me too’ drug development strategy is gradually shifting towards novel chemotypes and scaffolds. This approach may provide unique pharmacophores with better physicochemical and ADME properties, freedom-to-operate space and improved chemical expansion perspectives. It is exemplified by a dozen new kinase inhibitors currently being disclosed at the discovery stage or at preclinical development.
Most approved protein kinase inhibitors reversibly bind to protein pockets with ATP-competitive behaviour. Due to the fact that the ATP-binding cleft is highly conserved among members of the kinase family, the design of selective type I inhibitors is a challenge. Moreover, the compounds need to compete with high cellular ATP levels which leads to reduced efficacy and obligation for increased drug doses. Both issues potentially open the gates for toxicity in vivo.
Initially, type II inhibitors targeting the inactive kinase state were announced as a solution[xxvii],[xxviii]. This dogma no longer holds true[xxix]; for example, B-RAF selective type I inhibitor vemurafenib (Zelboraf®) presents a superior selectivity profile over sorafenib (Nexavar®), regorafenib (Stivarga®) and ponatinib (Iclusig®), all of which are type II inhibitors[xxx]. The selectivity data for the first kinase inhibitors with the type III binding mode (trametinib; MEK1 and MEK2) or type V (mixed profile of type I and II represented by lenvatinib) are more encouraging. However, whether the allosteric approach is key to selectivity success still remains to be elucidated. The recent approval of ibrutinib (BTK inhibitor) and afatinib (EGFR, Her2 inhibitor) able to covalently modify cysteine residues of their protein substrates has opened new perspectives for the development of more specific, targeted protein kinase inhibitors.
Better kinase selectivity improves tolerability with safer options for stand-alone or combination therapies. However, the paradigm of absolute selectivity required for kinase inhibitors is not critical nowadays, as long as the inhibitors have well balanced modulation properties towards the desired proteins and acceptable side effects in routine patient management.
Novel, selective kinase inhibitors have resulted in a number of breakthrough medicines for genetically well-defined populations. The current kinase inhibitors landscape highlights a need for exploring new avenues, innovative hypotheses and new indications not only in immunoncology areas but also beyond cancer. The latest achievements in the field have facilitated patients’ treatment benefits and created even more demanding expectations, hopes and promises for the future. Kinase inhibitors have yet to show their full potential.
References
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144(5):646-674
- Lahiry P, Torkamani A, Schork NJ, Hegele RA. Kinase mutations in human disease: interpreting genotype–phenotype relationships. Nature Reviews Genetics 2010;11(1):60–74
- Rask-Andersen M, Zhang J, Fabbro D, Schiöth HB. Advances in kinase targeting: current clinical use and clinical trials. Trends in Pharmacological Sciences. 2014;35(11):604-620
- Rajakulendran T, Sicheri Allosteric protein kinase regulation by pseudokinases: insights from STRAD. Science Signaling. 2010;3(111), pe8
- Shaw AS, Kornev AP, Hu J, Ahuja LG, Taylor Kinases and pseudokinases: lessons from RAF. Molecular and Cellular Biology. 2014;34:1538-1546
- Boudeau J, Miranda-Saavedra D, Barton GJ, Alessi DR. Emerging roles of pseudokinases. TRENDS in Cell Biology. 2006;16(9):443-452
- Kanner SB, Perez-Villar Altering T-cell activation by targeting the multidomain tyrosine kinase Itk. Trends in Immunology. 2003;24(5):249–253
- Sanjuan MA, Pradet-Balade B, Jones DR, Martinez-AC, Stine JC, Garcia-Sanz JA, Merida I.T cell activation in vivo targets diacylglycerol kinase α to the membrane: A novel mechanism of Ras attenuation. Journal of Immunology. 2003;170:2877-2883
- Sawasdikosol S, Zha R, Yang B, Burakoff S. HPK1 as a novel target for cancer immunotherapy. Immunol Res. 2012;54(1-3):262-5
- Alzabin S, Bhardwaj N, Kiefer F, Sawasdikosol S, Burakoff S. Hematopoietic progenitor kinase 1 is a negative regulator of dendritic cell activation. J Immunol. 2009;182(10):6187-94
- Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30(6):832–844
- Kang J, Huddleston SJ, Fraser JM, Khoruts A. De novo induction of antigen-specific CD4+CD25+Foxp3+ regulatory T cells in vivo following systemic antigen administration accompanied by blockade of mTOR. J. Leukoc. Biol. 2008;83(50):1230–1239
- Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, Rowan WC, Sancho S, Walker LS, Vanhaesebroeck B, Okkenhaug K. Cutting edge: the phospho-inositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+regulatory T cells. J Immunol. 2006;177(10):6598–6602
- Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H, Friedman L, Turner M, Okkenhaug K, Vanhaesebroeck B. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510(7505):407–411
- Yaqub S, Taskén K. Role for the cAMP-protein kinase A signaling pathway in suppression of antitumor immune responses by regulatory T cells. Crit Rev Oncog. 2008;14(1):57–77
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, Setty M, Leslie CS, Oei Y, Pedraza A, Zhang J, Brennan CW, Sutton JC, Holland EC, Daniel D, Joyce JJ. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272
- Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, Wang-Giliam A, Goedegebuure SP, Linehan DC, DeNardo DG. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–5069
- Mok S, Koya RC, Tsui C, Xu J, Rober L, Wu L, Graeber TG, West BL, Bollag G, Ribas A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014;74(1):153–161
- Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, de Gramont A. Targeting the TGFβ pathway for cancer therapy. Pharmacology & Therapeutics. 2015;147:22–31
- Wang SK, Chang RT. An emerging treatment option for glaucoma: Rho kinase inhibitors. Clin. Ophthalmol. 2014;8:883–890
- Wang G, Pan J, Chen S-D.Kinases and kinase signaling pathways: potential therapeutic targets in Parkinson’s disease. Prog. Neurobiol. 2012;98:207–221
- Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yanf Q, Lee J-D, Patricelli MP, Nomanbhoy TK, Alessi D, Gray N. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 2011;7:203–205
- Wang LH, Johnson EM. Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology. 2008;71:462
- Blalock EM, Geddes JW, Chen KC, Porter NM, Maresbery WR, Landfield PW. Incipient Alzheimer’s disease: microarray correlation analysis reveal major transcriptional and tumor suppressor responses. Proc. Natl. Acad, Sci. USA. 2004;101:2173-2178
- Fajardo RJ, Manoharan RK, Pearsall RS, Davies T, Monnel TE, Ucran JA, Pearsall AE, Khanzode D, Kumar R, Underwood KW, Roberts B, Seehra J, Bouxsein ML. Treatment with a soluble receptor for activin improves bone mass and structure in the axial and appendicular skeleton of female cynomolgus macaques (Macaca fascicularis). Bone. 2010; 46:64–71
- Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nature Biotech. 2011;29:1039–1045
- Davis M, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nature Biotech. 2011;29:1046–1051
- Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu Q, Gray NS. Exploration of type II binding mode: a privileged approach for kinase inhibitor focused drug discovery? ACS Chem. Biol. 2014;9:1230-1241
- Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discovery Today. 2016;21(1):5-10
Biographies
 Dr. Monika Dobrzańska holds a PhD degree in Medicinal Chemistry and MSc, Eng. in Biotechnology, specialising in Biocatalysis and Molecular Biotechnology from Wrocław University of Technology, Poland. Further to that, she completed a postgraduate studies Monitoring of Clinical Trials at Kozminsky University, Warsaw. In 2010, Dr. Dobrzanska joined Selvita, the largest drug discovery company in Central and Eastern Europe, engaged in the research and development of breakthrough therapies in the area of oncology. Since the beginning of her professional career, Dr Dobrzańska has been involved in strategic development of medicinal chemistry for kinase inhibitors in the preclinical phase projects. Currently, she is responsible for multidisciplinary project management in oncology and immunology.
Dr. Monika Dobrzańska holds a PhD degree in Medicinal Chemistry and MSc, Eng. in Biotechnology, specialising in Biocatalysis and Molecular Biotechnology from Wrocław University of Technology, Poland. Further to that, she completed a postgraduate studies Monitoring of Clinical Trials at Kozminsky University, Warsaw. In 2010, Dr. Dobrzanska joined Selvita, the largest drug discovery company in Central and Eastern Europe, engaged in the research and development of breakthrough therapies in the area of oncology. Since the beginning of her professional career, Dr Dobrzańska has been involved in strategic development of medicinal chemistry for kinase inhibitors in the preclinical phase projects. Currently, she is responsible for multidisciplinary project management in oncology and immunology.
 Dr. Karolina Gluza received her PhD in Chemistry from Wroclaw University of Technology, investigating the mechanism of the complexation reaction between diols and boronic acids using computational chemistry methods. Since November 2013 she has been a member of the computer-aided drug design group at Selvita. She works as Senior Scientist and is mainly involved in development of small molecule inhibitors of kinases for anticancer therapy.
Dr. Karolina Gluza received her PhD in Chemistry from Wroclaw University of Technology, investigating the mechanism of the complexation reaction between diols and boronic acids using computational chemistry methods. Since November 2013 she has been a member of the computer-aided drug design group at Selvita. She works as Senior Scientist and is mainly involved in development of small molecule inhibitors of kinases for anticancer therapy.
Related topics
Kinases
Related conditions
Leukaemia
Related organisations
Selvita
Related people
Karolina Gluza, Monika Dobrzańska