Using native mass spectrometry to inform drug discovery
Posted: 2 June 2017 | Carol V Robinson, Hsin-Yung Yen, Idlir Liko, Jonathan ST Hopper, Timothy Allison | No comments yet
Mass spectrometry (MS) performed under native-like conditions (nMS) is a promising approach for studying protein-drug interactions, and has already informed treatments of some of the most intractable diseases including cancer, diabetes and Alzheimer’s.

At the core of nMS is the ability to preserve non-covalent interactions and perform very accurate mass measurements. This ability has enabled advances in structural biology, including the elucidation of protein homogeneity, oligomeric state, sequence variation and the identity of bound ligands. These advances are made possible by preserving proteins in their native state in solution, followed by careful transfer into the gas phase of the mass spectrometer. As a consequence, nMS is finding applications in increasingly diverse research areas such as high-throughput drug screening, the study of amyloid formation and inhibition, the characterisation of antibody-drug conjugates and the elucidation of the interactions of membrane proteins with lipids and therapeutics. This ability to study the influence of small molecule ligand binding on the cascade of protein interactions that underlie many disease states is therefore providing a new paradigm in drug discovery.
A better understanding of cellular processes is of paramount importance for the development of new therapeutics. These processes are enabled by synchronisation of multiple protein-protein or protein-ligand interactions and, while they dictate how cells interact in tissues, they are guided by molecular rearrangement at the atomic level. Hence, new technologies that are capable of capturing whole protein complexes, yet providing atomic-level details for their structure and function, hold the potential to unravel the complexity of these interactions. Over the last decade nMS in combination with ion mobility (IM) – which measures the overall conformation of an ion – has emerged as a promising method to study protein interactions at equilibrium.
nMS begins with the isolation of protein complexes from their cellular environment and is followed by exchanging the analyte to an MS-compatible medium, while preserving the protein in its folded, native state. Protein complexes are then ionised and transferred into the gas phase via soft ionisation methods such as electrospray ionisation. By optimising the instrumental parameters, ions are desolvated while their folded structure and non-covalent interactions are maintained. This allows the mass of protein and their bound ligands to be determined with high accuracy and resolution, elucidating not only their binding stoichiometry but also the capability to observe each species present in solution. This is a powerful attribute, since most biophysical methods report only the average ensemble present in solution.
Once inside the mass spectrometer, individual protein-ligand complexes can be isolated and dissociated to provide ligand identification or to quantify the stabilisation that ligands provide upon binding to the proteins.^1 The latter can be achieved by monitoring how proteins unfold in the presence or absence of bound ligands. This experiment is performed in combination with IM, a method that measures the time it takes for ions to pass through a tube filled with inert gas known as a drift tube. This drift time is dependent on a protein’s charge and rotational average collision cross-section, or in more simplistic terms – its globular shape. Utilising this additional dimension of separation, it is possible to differentiate highly complex mixtures composed of species of equivalent mass, known as isobaric species, based on different conformations.
In addition, by increasing the activation in the collision cell, proteins can also be fragmented to provide information regarding a primary sequence and report the presence of post-translational modifications, typically known as top-down proteomics. The information derived from nMS can also be combined with results obtained from x-ray crystallography or electron microscopy to refine the protein structures by reporting the identity of bound ligands that reside in areas with weaker electron density.^2
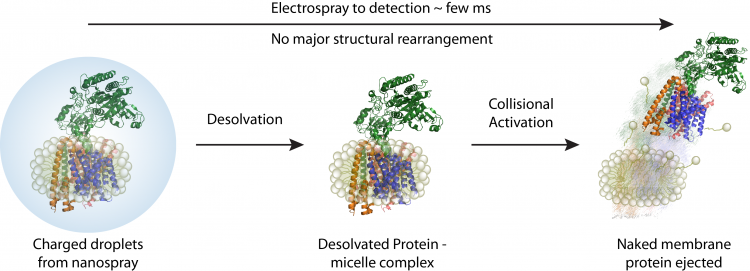
A schematic depicting how membrane proteins are studied using native mass spectrometer. Initially proteomicelles are ionised and desolvated , subsequently the detergent is removed inside the mass spectrometer
While nMS has predominantly been applied by academic researchers, its exciting capabilities are now proving useful in the pharmaceutical industry where elucidation of specific small-molecules or biologic interactions with drug targets is of growing importance. As pharmaceutical scientists take on increasingly complex and more challenging novel targets – often involving more than one protein in a complex – the ability of nMS to inform on the binding, the conformational changes and strength of the binding, can be a powerful tool to advance drug discovery efforts. In this review, we highlight recent applications where native MS has proved critical for target characterisation or for defining interactions with small molecules or biologics.
Direct visualisation of drug binding
The ability to maintain non-covalent interactions is best exemplified by the preservation of protein-ligand interactions. This has allowed nMS to be directly employed in the identification of compounds that serve as a starting point for drug discovery, especially via fragment-based drug discovery. This approach involves screening a drug library composed of low-molecular-weight drug fragments, and identifying multiple fragments that bind to the target. The bound fragments can then be either chemically extended or linked together to achieve a highly selective drug.
Since a fragment typically forms only a few interactions with target proteins, their binding is often weak; nevertheless, native mass spectrometry can capture interactions that have dissociation constants in the mM to µM range. Using a miniaturised electrospray ionisation platform and automated systems, hundreds of fragments can be screened with low sample and time consumption.^3 The results obtained by nMS are in good agreement with similar results obtained from nuclear magnetic resonance, X-ray crystallography and surface plasmon resonance.^4 Furthermore, nMS can perform simultaneous ligand competition assays on a number of drugs introduced in the same solution that target a particular protein. The resulting mass shifts, due to ligand binding, can be identified by subsequent fragmentation.
This method provides a means of performing ligand competition assays and highlights drugs that bind with the highest affinity. It is also possible to capture synergistic effects – or cooperatively of multiple drug/cofactor binding events. Once a particular hit is identified, nMS can also be employed to determine the dissociation constant of each ligand.^5 Given its large mass range, analogous approaches can be used to measure large protein assembles, in addition to small molecules. nMS can also therefore elucidate the identity of peptide-like drugs that target biologics including intact antibodies (see below).
Studying amyloid formation
A variety of neurodegenerative disorders, such as Parkinson’s and Alzheimer’s, are associated with the formation of amyloids. Whilst the implication of amyloidosis in the deceased is yet to be elucidated, there is emerging evidence that early-stage intermediates are responsible for the development of the disease.^6 To study these intermediates, however, is highly challenging due to their inherent ability to aggregate. Finding conditions to initially preserve monomeric species and then nucleate the reaction to form higher order oligomers, allows controlled conditions to monitor the time-dependent formation of early stage intermediates by native mass spectrometry.
By taking samples from healthy and diseased organisms, and using nMS to analyse all species present in solution instantaneously, it was found that monomeric units are present in multiple conformations, but only one particular conformation could be directly linked to the disease state. For instance, in the case of transthyretin, dimers and tetramers are more stable relative to other intermediates that quickly aggregate.^7 High-resolution mass measurements also allow researchers to elucidate the importance of primary sequences in fibroid formation. For example, full-length monomers of Aβ aggregate significantly faster than a truncated variant from which two amino acids have been removed.^8 Furthermore, IM of both full-length and truncated variants showed that they assemble via two different mechanisms. In the first, by an open tetramer that allows for formation of high-order oligomers, and in the second, a compact one where there is no place for other monomeric proteins to attach. Being able to capture the early stages of amyloid formation can be used to test for drug treatments that reverse this process.^9
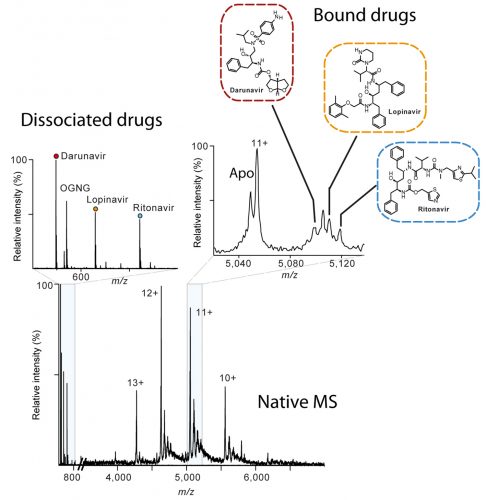
Native mass spectrum of the human metalloprotease ZMPSTE24 bound to lopinavir, ritonavir and darunavir. The inset on the left shows the presence of the drugs in solution whereas the zoom spectrum at charge 11+ shows how that all three drugs are bound to the protein conforming the alibity of nMS for off-target drug binding.
An added dimension in antibody characterisation
Pharmaceutical companies are harnessing proteins, such as antibodies, to act as therapeutic agents by binding to target antigens. Although these therapeutic proteins possess remarkable abilities, their protein structure and modifications must be controlled and regulated with high precision in order to produce consistent results. Initial nMS studies on antibody-antigen complexes determined the stoichiometry of antigens bound to antibodies, but were unable to observe the myriad of modifications – mainly glycosylation on the antibodies themselves.^10
Recent developments in increasing the resolution of mass spectrometers allow the many glycoforms present in antibodies to be separated, as shown for the best-selling biotherapeutic Herceptin used to treat breast cancer, oesophageal cancer and stomach cancer.^11 Combined with conventional proteomics methods, nMS provides a full characterisation of antibody post-translational modifications, reporting on glycan quantitation and characterisation as well as glycoprotein occupancy, and can be directly used to monitor the production of antibodies with a specific modification. Of high importance is the ability of nMS to monitor reactions where therapeutics are attached to a specific part of the antibody. While varying multiple coupling conditions, the mass of each species present in solution can be recorded, thus finding conditions where a high percentage of the products are favoured and enabling the use of nMS to guide the production antibody-drug conjugates.
Elucidating the interaction of membrane proteins with lipids and drugs
Given that proteins can exist in multiple structures and engage with different binding partners in response to environmental change, their dynamics are notoriously difficult to characterise. Moreover, a third of all proteins reside in the ‘greasy’ walls of cells and are known as membrane proteins. Since most biophysical processes require proteins to first be dissolved in aqueous environments, membrane proteins present significant technical challenges due to their hydrophobic nature. Furthermore, two-thirds of current drugs target membrane proteins, so it is easy to understand the frustration of working with these targets and the importance of overcoming these challenges.
It was only during the last decade that the difficulties of carrying out nMS experiments with membrane proteins were surmounted. Early research into these complexes uncovered the relationship between lipids and membrane proteins in rotary ATPases, large 0.7 MDa enzymes that synthesise ATP – the energy currency of all cells – and the target of a number of antimicrobial agents. This was significant not only since close to 30 subunits remained intact in vacuum, but also because it uncovered new mechanistic insight.^12 These experiments were then followed with the development of protocols where nMS can be used to monitor the extent of delipidation and to identity structurally important lipids. Once this was achieved, and conditions where only important lipids were retained occurred, the focus changed to drug binding using, as first example, ABC transporters – proteins involved in multi-drug resistance and cancer. When coupling with IM MS it was shown that lipids and drugs act synergistically to promote drug efflux of cancer therapeutics through the P-glycoprotein pump.^13
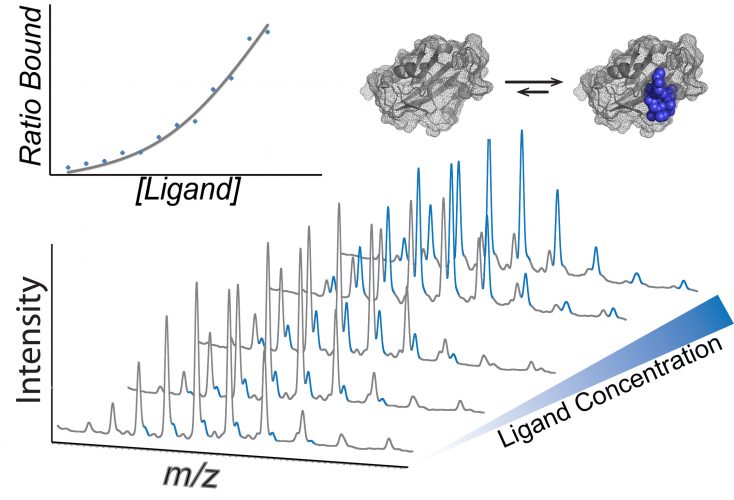
Mass spectra showing how dissociation constants can be calculated using native mass spectrometry. A concertation series of the ligand is incubated with the protein of interest and the intensity of each species is used to calculate the KD (inset).
As well as uncovering the beneficial mechanisms of drugs, mass spectrometry is also able to capture drugs bound to proteins, including ones that cause devastating side effects. For example, while studying the interactions of the drugs lopinavir and ritonavir, intended for HIV therapy, nMS was used to show how they blocked a critical enzyme pathway linked to progeria-like symptoms which cause premature aging.^14
More recently, researchers at OMass Technologies have been applying nMS to study G-protein coupled receptors (GPCRs) – perhaps one of the most challenging targets to date. Finding mixed micelle conditions that preserve the native state of these notoriously dynamic molecules required an exhaustive screen of nMS and solution conditions. These studies are already providing new insight, showing how drugs compete with natural ligands for binding to orthosteric sites and have highlighted the effects of drug binding on posttranslational modifications.^15 Very recently, this research has uncovered new roles for lipids in stabilising receptors and highlighted new opportunities for drug discovery at these lipid binding sites.
During the last 20 years, native MS has provided key information on some of the most difficult disease targets in biomedical research. Many biotech and pharmaceutical companies such as OMass technologies, UCB, Amgen and Genentech, have integrated nMS into their discovery pipelines, focussing on new membrane targets and biologics. We are confident that this exciting technology will therefore play a major role in the discovery of the next generation of therapeutics – drugs or proteins – that target diseases of the 21st century.
Acknowledgements
We acknowledge funding from the ERC (ENABLE), the MRC (G1000819) and a Wellcome Trust Investigator Award (104633/Z/14/Z). We thank David Tickle at MRC Technology, Mark Agasid and Weston Struwe, at the University of Oxford for helpful discussions, and Carina Monico for taking the photographs.
Biographies
 Idlir Liko is currently the Chief Technology Officer at OMass Technologies. He holds a master’s degree (MSc) form ETH Zurich and a DPhil from the University of Oxford, supervised by Prof Robinson, where he focussed on the effect ligands and PTMs have on the structure and stability of membrane proteins, once they are transferred into the gas-phase. In 2016, he co-founded OMass Technologies, a company specialised in native mass spectrometry of membrane proteins.
Idlir Liko is currently the Chief Technology Officer at OMass Technologies. He holds a master’s degree (MSc) form ETH Zurich and a DPhil from the University of Oxford, supervised by Prof Robinson, where he focussed on the effect ligands and PTMs have on the structure and stability of membrane proteins, once they are transferred into the gas-phase. In 2016, he co-founded OMass Technologies, a company specialised in native mass spectrometry of membrane proteins.
 Timothy M Allison has a background in protein engineering and completed his PhD in biochemistry at the University of Canterbury (New Zealand). Over the past five years he helped to extend native mass spectrometry to the study of membrane proteins. He currently works in the laboratory of Professor Dame Carol Robinson and advises OMass Technologies in his role as a scientific consultant.
Timothy M Allison has a background in protein engineering and completed his PhD in biochemistry at the University of Canterbury (New Zealand). Over the past five years he helped to extend native mass spectrometry to the study of membrane proteins. He currently works in the laboratory of Professor Dame Carol Robinson and advises OMass Technologies in his role as a scientific consultant.
 Hsin-Yung Yen has been a postdoctoral researcher from the Professor Dame Carol Robinson research group in the Department of Chemistry, University of Oxford since 2014. He is devoting his time to studying protein biology by means of native mass spectrometry, especially focussing on the lipid dynamics of membrane proteins and its functional relation to GPCRs.
Hsin-Yung Yen has been a postdoctoral researcher from the Professor Dame Carol Robinson research group in the Department of Chemistry, University of Oxford since 2014. He is devoting his time to studying protein biology by means of native mass spectrometry, especially focussing on the lipid dynamics of membrane proteins and its functional relation to GPCRs.
 Jonathan T.S. Hopper completed his PhD in fundamentals of native mass spectrometry at the University of Nottingham, during which time he gained experience working in analytical research teams within the pharmaceutical sector. He then became a postdoctoral research associate at the University of Oxford. As CEO of OMass Technologies, a company he co-founded, he is now committed to delivering native mass spectrometry technology to drive pharmaceutical research.
Jonathan T.S. Hopper completed his PhD in fundamentals of native mass spectrometry at the University of Nottingham, during which time he gained experience working in analytical research teams within the pharmaceutical sector. He then became a postdoctoral research associate at the University of Oxford. As CEO of OMass Technologies, a company he co-founded, he is now committed to delivering native mass spectrometry technology to drive pharmaceutical research.
 Professor Dame Carol Robinson holds the Chair of Dr Lee’s Professor of Chemistry at the University of Oxford. She is recognised for using mass spectrometry to further her research into the 3D structure of proteins and their complexes. Recent highlights from her work include the discovery that membrane protein complexes can be liberated from micelles in the gas phase while retaining their subunits interactions, lipid binding properties and overall topology. In 2016 she co-founded OMass technologies. Prof Robinson is a Fellow of the Royal Society and was this year elected as a foreign associate to the National Academy of Sciences USA.
Professor Dame Carol Robinson holds the Chair of Dr Lee’s Professor of Chemistry at the University of Oxford. She is recognised for using mass spectrometry to further her research into the 3D structure of proteins and their complexes. Recent highlights from her work include the discovery that membrane protein complexes can be liberated from micelles in the gas phase while retaining their subunits interactions, lipid binding properties and overall topology. In 2016 she co-founded OMass technologies. Prof Robinson is a Fellow of the Royal Society and was this year elected as a foreign associate to the National Academy of Sciences USA.
References
- Allison TM, Reading E, Liko I, Baldwin AJ, Laganowsky A, Robinson CV. Quantifying the stabilizing effects of protein-ligand interactions in the gas phase. Nat Commun. 2015;6.
- Liko I, Allison TM, Hopper JT, Robinson CV. Mass spectrometry guided structural biology. Curr Opin Struct Biol. 2016;40:136-44.
- Maple HJ, Garlish RA, Rigau-Roca L, Porter J, Whitcombe I, Prosser CE, et al. Automated protein–ligand interaction screening by mass spectrometry. J Med Chem. 2012;55(2):837-51.
- Woods LA, Dolezal O, Ren B, Ryan JH, Peat TS, Poulsen S-A. Native state mass spectrometry, surface plasmon resonance, and X-ray crystallography correlate strongly as a fragment screening combination. J Med Chem. 2016;59(5):2192-204.
- Hopper JTS, Robinson CV. Mass spectrometry quantifies protein interactions—from molecular chaperones to membrane porins. Angew Chem Int Ed. 2014;53(51):14002-15.
- Kotler SA, Walsh P, Brender JR, Ramamoorthy A. Differences between amyloid-β aggregation in solution and on the membrane: insights into elucidation of the mechanistic details of Alzheimer’s disease. Chem Soc Rev. 2014;43(19):6692-700.
- Cole HL, Kalapothakis J, Bennett G, Barran PE, MacPhee CE. Characterizing early aggregates formed by an amyloidogenic peptide by mass spectrometry. Angew Chem. 2010;122(49):9638-41.
- Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nature chemistry. 2009;1(4):326-31.
- Young LM, Saunders JC, Mahood RA, Revill CH, Foster RJ, Tu L-H, et al. Screening and classifying small-molecule inhibitors of amyloid formation using ion mobility spectrometry–mass spectrometry. Nature chemistry. 2015;7(1):73-81.
- Tito MA, Miller J, Walker N, Griffin KF, Williamson ED, Despeyroux-Hill D, et al. Probing molecular interactions in intact antibody: antigen complexes, an electrospray time-of-flight mass spectrometry approach. Biophys J. 2001;81(6):3503-9.
- Parsons TB, Struwe WB, Gault J, Yamamoto K, Taylor TA, Raj R, et al. Optimal synthetic glycosylation of a therapeutic antibody. Angew Chem Int Ed. 2016;55(7):2361-7.
- Zhou M, Morgner N, Barrera NP, Politis A, Isaacson SC, Matak-Vinkovic D, et al. Mass spectrometry of intact V-type ATPases reveals bound lipids and the effects of nucleotide binding. Science. 2011;334(6054):380-5.
- Marcoux J, Wang SC, Politis A, Reading E, Ma J, Biggin PC, et al. Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc Natl Acad Sci USA. 2013;110(24):9704-9.
- Mehmood S, Marcoux J, Gault J, Quigley A, Michaelis S, Young SG, et al. Mass spectrometry captures off-target drug binding and provides mechanistic insights into the human metalloprotease ZMPSTE24. Nature Chemistry. 2016.
- Yen H, Hopper JTS, Liko I, Zhu Y, Wang D, Stegmann M, et al. Ligand binding to a G-protein coupled receptor captured in a mass spectrometer. Science Advances . 2017;in press
Related topics
Mass Spectrometry
Related organisations
OMass Technologies, Oxford University