Practical application of in vivo optical imaging in biotherapeutic biodistribution analyses
Posted: 14 September 2016 | Dr Norman Peterson (MedImmune/AstraZeneca) | No comments yet
As advances in protein engineering technology have led to the production of novel and more structurally complex biopharmaceuticals, the ability of these drugs to smoothly traverse the body and arrive at their intended target site has become more unpredictable…
These advances have provided newly designed antibody-based therapeutics that have multi-specific binding properties and functions, novel linkers, and carry that cytotoxic payloads (antibody-drug conjugates). Many of these molecules have poor pharmacokinetics, due to either immunogenicity and/or poorly understood mechanisms of tissue processing and clearance. With regard to the latter, the biotherapeutic may be retained, bound and/or delayed at the tissues that are actively or passively/nonspecifically responsible for their removal from the systemic circulation.
Fc receptors in the liver, skin and kidneys are partially responsible for clearance (and recycling) of antibody-based therapeutics1, and recently the liver sinusoidal endothelial cells (LESC) have been implicated as an additional mechanism2. Now, with the help of identifiable tags and non-invasive molecular imaging modalities, the fate of these biotherapeutics can be monitored in the body in real time.
Whereas pharmacokinetic studies provide information on whether biotherapeutics are rapidly cleared, in vivo imaging can be used to elucidate the percentage of a given quantity of labelled biotherapeutic injected into an animal that is getting both to the target site and unintended sites. The ability to gather biodistribution information in preclinical studies noninvasively has evolved over the past 25 years as several of the modalities used in the clinic, such as ultrasound, positron emission tomography (PET) and magnetic resonance imaging (MRI) have been adapted for use with animal models. Additionally, improvements in labels that are readily conjugated and produce stronger signals have improved versatility. Computational algorithms to correct for noise and distortions have also improved making resolution and location of previously ambiguous signals more defined. Lastly, more user-friendly software analysis programmes have allowed access with less user training.
Labelling types
Although there are multiple methods for analysing the biodistribution of labelled biotherapeutics in vivo, the two most commonly used preclinical methods employ radioisotope labelling with PET and near-infrared fluorescence labelling with optical imaging. Based upon publication trends, both modalities have recently experienced increased application with optical imaging passing PET in the past four years (Figure 1).
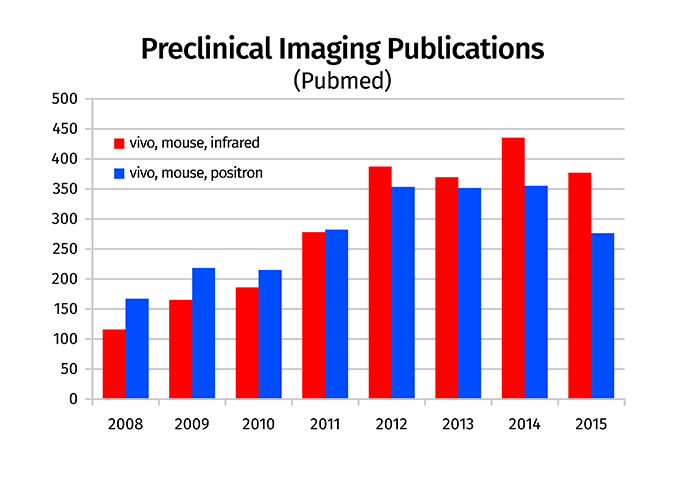
Figure 1: Trends in imaging modality publications. Literature searches in Pubmed were performed using keywords ‘mouse, vivo, positron’ and ‘mouse, vivo, infrared’ to estimate trends in the use of PET and optical imaging, respectively
The increasing popularity of optical imaging can be partially attributed to the availability of multiple near-infrared fluorescent labels which allow deeper tissue penetration than their predecessors that have shorter wavelength emissions, as well as innovative approaches for clinical application, and an increase in constructs containing both fluorescence and radioisotope labels.
Additionally, optical imaging is less costly and much simpler to use in terms of data analysis and does not have to comply with the technical details associated with radioisotopes. However, because of its relatively high sensitivity and resolution, PET/CT remains the industry gold standard for in vivo biodistribution studies. Because of its limited tissue penetrance, in vivo biodistribution analyses with optical imaging are not possible in species larger than mice. If there is support and infrastructure available, having both optical imaging and PET/CT provides a versatile and highly applicable resource for any drug development programme.
Dual imaging research
We have utilised two optical imaging modalities in over 50 rodent studies at our institution: a multi-spectrum fluorescence/bioluminescence imager (MSFI) and a fluorescence tomography system (FTS). The main difference between the two is that the FTS uses a moving laser to excite the fluorophore and absorbed/emitted light is detected to create a three-dimensional reconstruction of the label’s location within the body, whereas in the MSFI the excitation light is produced by a diffuse light source.
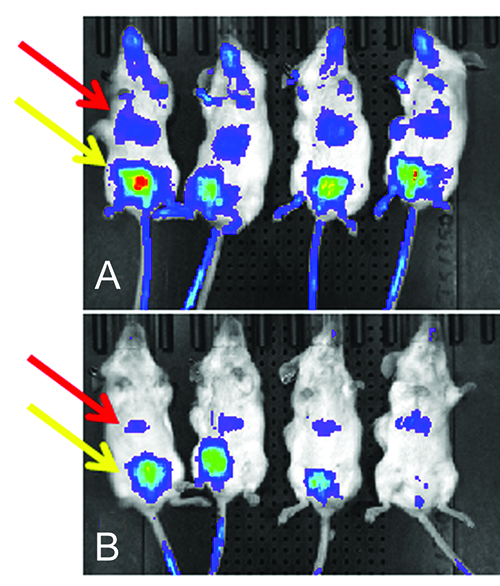
Figure 2: Surface fluorescence from mice injected with labelled antibodies. MSFI from mice 48 hours after injection of two lead candidate antibodies labelled with Alexa Fluor 750. Candidate A had greater fluorescence from the area above the liver and faster clearance from the blood than candidate B. Red arrow denotes fluorescence from the liver and yellow area denotes fluorescence from the bladder
Initially, the FTS body scans from the thorax to the lower abdomen were used in our biodistribution analyses, however this was found to be very time-consuming and reconstructions commonly failed, presumably due to lack of signal detection and/or adequate contrast of signal throughout the tissues. In our experience the FTS is better suited for analyses of deep tissues within an area of 20mm3 or less. The MSFI has the advantage that the entire surface area of five mice can be imaged simultaneously and signal in subcutaneous tumours can be readily detected.
The major drawback of conducting biodistribution analysis of biotherapeutics with MSFI is the limited tissue penetrance of light. In several cases, however, we were able to detect retention of NIRF-labelled antibodies in vivo in the livers of mice. In antibodies with poor PK, the liver was commonly identified as a site of NIRF-labelled antibody retention within the first 24 hours of administration (Figure 2). NIRF signal from the kidneys may also penetrate through the body when the mouse is positioned in either the lateral or dorso-ventral orientation.
Interpretation of fluorescence from the kidney may be difficult as it may be the result of specific binding, nonspecific retention and/or an accumulation of metabolic breakdown products. With respect to the latter, we commonly see increased fluorescence from the kidneys and/or bladder associated with elevated liver fluorescence. Hypothetically, this may be the result of metabolic breakdown at the liver with incomplete elimination through the biliary system and leakiness back into the circulation, to ultimately be cleared by the kidneys.
Owing to the fact that solid tumours concentrate and form well-defined targets, the majority of applications for in vivo imaging have been in oncology. MSFI works well in nude mouse subcutaneous tumour xenograft models that have been intravenously injected with fluorescently-labelled antibodies. Imaging should be performed when tumours are growing in log phase (~ 500-1000mm3) to avoid necrosis and/or ulceration. Specificity can be demonstrated by a few different methods: test antibodies can be compared with a similarly labelled control antibody; or by competition with 10-20 fold excess unlabelled antibody injected 30-60 minutes prior to injection of the labelled antibody. In the competition analysis, the addition of unlabelled antibody should result in a decrease in fluorescence from the tumour, unless there is a large off-target tissue that is retaining the antibodies. Thirdly, fluorescence from tumours expressing and not expressing the target can be compared. These tumours can be implanted in separate mice or both implanted on opposite flanks of the same mouse. The advantage of the latter is that few mice are needed and intra-animal variability is reduced. The disadvantages are that tumour growth characteristics should be well characterised so that implantation of each can be timed such that sizes are equal at imaging. Additionally, one should be aware of the potential for each tumour to affect the microenvironment of its opposite tumour.
At the end of each study, typically 48-72 hours post-administration, ex vivo analysis of tissues (and tumour) is performed to confirm in vivo results and to detect any signal that may not have been strong enough to penetrate the body cavity. These ex vivo analyses are particularly useful for analysing skin sections. We have found that, second to the liver, the skin is a site of nonspecific antibody retention. Dendritic cell binding and a relatively high concentration of Fc receptors in the skin likely contributes to this observation3. For increased sensitivity and comparative analyses of fluorescence in the lungs and spleen, it may be necessary to include additional time-point cohorts for ex vivo analyses of these tissues. However, the need for additional groups in this case negates the advantage of being able to perform longitudinal studies with in vivo imaging. This would likely be unnecessary with application of the more sensitive PET.
Different intrinsic properties of the labels can also influence results and this is true for both fluorophores and radioisotopes. Threshold settings at the longer wavelengths tend to pick up less background from tissue autofluorescence, especially from the stomach, thus fluorophores with wavelengths >700nm are preferred. We and others have found that some fluorophores have longer tissue retention than others and this is most prominent in the liver4,5. Longer tissue retention can be advantageous in allowing signal to accumulate and intensify for improved detection. Because multiple fluorescent labels can be administered simultaneously, or in conjunction with bioluminescence in the same animal, studies with different proteins or biomarkers of interest can be performed and offer the ability to determine colocalisation of multiple markers; this is not possible with PET.
Most MSFI have the ability to measure bioluminescence as well as fluorescence and when used with mice containing luciferase-induced reporter constructs it can be a useful model for assessing drug pharmacodynamics. Various mouse models with cytokine, cell marker or signal transduction pathway-induced luciferase reporters have been developed. Because expression variability and background is high in these models, baseline bioluminescence analysis is needed prior to post-treatment imaging. This technology can also be used to track tumour cells, bacteria and viruses which express the luciferase transgene and there is no background emission from wild type mice that could obscure signal detection. In summary, with an MSFI platform, studies can be designed with fluorescence tagging of biotherapeutics to not only monitor where they are retained, but also determine where they elicit an effect via induction of luciferase-reporter transgenes.
Imaging evolution
The field of in vivo imaging for preclinical studies is constantly evolving. One of the drawbacks of several of the modalities being used today for imaging biodistribution is that they lack the ability to sufficiently define the soft tissue microenvironment or tissue borders of the target signal. To this end, combined PET-MRI systems have recently become commercially available and the technology of this combined modality will likely be improved upon in the near future6. Multi-spectral opticacoustical (MSOT) tomography is another relatively new modality that touts increased sensitivity and resolution when compared with MSFI and FTS7. Similar to FTS, light in the near-infrared spectrum is projected into the mouse, but sound waves emitted from the excited fluorescent tags are detected. Within the same time frame the system can also leverage the autoflourescence information collected from haemoglobin and soft tissues to provide microenvironmental context. As is frequently alluded to, no imaging modality can meet all the needs of a research institution that engages in multiple areas of study. Imaging resource investments should be thought of as contributing to a tool box, with each tool being added according to priority and scope of applicability. As discussed in this article, in vivo optical imaging with fluorescence labelling offers a flexible and practical start upon which to build.
Biography
 DR NORMAN PETERSON received his BS, MS, and DVM from the University of Illinois. He completed his residency training in Laboratory Animal Medicine and obtained a PhD in Comparative Medicine from the University of Pennsylvania. He was on faculty at Johns Hopkins University prior to his current position as Director of Veterinary Sciences at MedImmune/AstraZeneca.
DR NORMAN PETERSON received his BS, MS, and DVM from the University of Illinois. He completed his residency training in Laboratory Animal Medicine and obtained a PhD in Comparative Medicine from the University of Pennsylvania. He was on faculty at Johns Hopkins University prior to his current position as Director of Veterinary Sciences at MedImmune/AstraZeneca.
References
- Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 2004 Nov; 93 (11): 2645-2668
- Datta-Mannan A, Croy JE, Schirtzinger L, Torgerson S, Breyer M, Wroblewski VJ. Aberrant bispecific antibody pharmacokinetics linked to liver sinusoidal endothelium clearance mechanism in cynomolgus monkeys. MAbs 2016 Jul; 8 (5): 969-982
- Borvak J, Richardson J, Medesan C, Antohe F, Radu C, Simionescu M, et al. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int Immunol 1998 Sep; 10 (9): 1289-1298
- Peterson NC, Wilson GG, Huang Q, Dimasi N, Sachsenmeier KF. Biodistribution Analyses of a Near-Infrared, Fluorescently Labeled, Bispecific Monoclonal Antibody Using Optical Imaging. Comp Med 2016; 66 (2): 90-99
- Conner KP, Rock BM, Kwon GK, Balthasar JP, Abuqayyas L, Wienkers LC, et al. Evaluation of near infrared fluorescent labeling of monoclonal antibodies as a tool for tissue distribution. Drug Metab Dispos 2014 Nov; 42 (11): 1906-1913
- Ko GB, Yoon HS, Kim KY, Lee MS, Yang BY, Jeong JM, et al. Simultaneous multi-parametric PET/MRI with silicon photomultiplier PET and ultra-high field MRI for small animal imaging. J Nucl Med 2016 Apr 14
- Egusquiaguirre SP, Beziere N, Pedraz JL, Hernandez RM, Ntziachristos V, Igartua M. Optoacoustic imaging enabled biodistribution study of cationic polymeric biodegradable nanoparticles. Contrast Media Mol Imaging 2015 Nov-Dec; 10 (6): 421-427
Related topics
Antibodies, Biopharmaceuticals, Imaging, In Vivo, Pharmacology, Positron emission tomography (PET), Protein, Therapeutics