A key player in brain communication and mood regulation, the pharmaceutical industry views the NMDAR as the central pillar for next-generation therapies for depression. Dirk Beher from FundaMental Pharma reveals new strategies for targeting this important receptor.
The N-methyl-D-aspartate receptor (NMDAR) is a critical ion channel expressed in neurons throughout the brain, which plays a central role in neuronal development, synaptic plasticity and the cognitive processes of learning and memory.1 Structurally, the receptor functions as a heterotetramer, an assembly of four subunits that typically includes two obligatory GluN1 subunits paired with two regulatory subunits. These are most commonly GluN2A or GluN2B, though GluN2C/D or GluN3A/B may appear depending on the brain region and developmental stage. Belonging to the family of receptors that respond to glutamate, the primary excitatory neurotransmitter in the brain, the NMDAR operates under a unique activation mechanism. At a resting membrane potential of approximately -70mV, the channel is blocked by a magnesium ion (Mg2+). Activation requires the simultaneous binding of glutamate and a co-agonist, such as glycine or D-serine, along with neuronal depolarisation to repel this magnesium block. This opening allows calcium ions (Ca2+) to flow into the neuron, triggering a chemical cascade that promotes neuronal survival and strengthens synaptic connections. This strengthening, known as long-term potentiation (LTP), serves as the biological basis for neuroplasticity and the ability to form new memories. Despite its vital role in cognition, NMDAR function represents a double-edged sword as it is also linked to excitotoxicity, a damaging process implicated in neurological conditions such as Alzheimer’s disease (AD) and stroke,2 positioning the NMDAR as a potential drug target for neurodegeneration.
Clinical translation: a history of failures and successes
Despite the link between NMDAR over-activation and excitotoxicity, early attempts to target this receptor for neuroprotection resulted in a series of high-profile clinical failures.3 In the 1990s and early 2000s, pharmaceutical companies developed high-affinity, competitive antagonists (such as D-APV) and uncompetitive blockers (such as MK-801) to treat acute ischemic stroke, where glutamate transport systems are heavily compromised, leading to excitotoxicity. While effective in animal models when used as prevention treatment, these drugs failed in human trials as intervention treatment – likely due to the narrow time window for neuroprotection. In addition, NMDAR antagonists often cause severe psychotomimetic side effects like hallucinations, agitation and coma. Retrospective analysis revealed one of the key problems with these drugs was indiscriminate blocking of all NMDARs, including the physiological, synaptic receptors required for neuronal survival and normal brain function.
Success in targeting the NMDAR was finally achieved by shifting the therapeutic strategy from indiscriminate ‘blocking’ to sophisticated ‘modulation’.
Success in targeting the NMDAR was finally achieved by shifting the therapeutic strategy from indiscriminate ‘blocking’ to sophisticated ‘modulation’. A pioneering example is memantine, approved in 2002 for moderate-to-severe AD.4 In contrast to the failed high-affinity blockers, memantine is a low-affinity, uncompetitive antagonist characterised by rapid ‘on-off’ kinetics. This unique property allows it to block the channel only during prolonged, pathological activation (excitotoxicity) while quickly detaching to permit normal physiological signals to pass – effectively distinguishing between pathological ‘noise’ and physiological ‘signal’. The second major breakthrough involves ketamine, originally developed as an anaesthetic in the 1960s. While racemic ketamine has been used off-label for depression, its S-enantiomer, esketamine, was formally approved in 2019 as a rapid-acting antidepressant for treatment-resistant depression (TRD).5 Diverging from traditional antidepressants that modulate monoamines, ketamine is believed to preferentially block NMDARs on inhibitory interneurons, thereby causing disinhibition of excitatory neuron networks.6 The disinhibition is thought to result in a paradoxical burst of glutamate release, which stimulates ionotropic AMPA-type glutamate receptors and triggers downstream signalling pathways leading to rapid synaptic plasticity (synaptogenesis).7 Most recently, the validation of NMDAR modulation extended beyond TRD to major depressive disorder (MDD) with the approval of Auvelity (a combination of dextromethorphan and bupropion).8 Dextromethorphan acts as a low-affinity, uncompetitive NMDAR antagonist and when combined with bupropion to inhibit its rapid metabolism, it achieves sustained therapeutic levels in the brain.
Collectively, these successes demonstrate that the NMDAR remains a highly viable drug target, provided the therapeutic mechanism respects the delicate balance between preventing excitotoxicity and preserving necessary neuroplasticity.9
Esketamine and the paradigm shift: from monoamines to glutamate
The approval of Spravato® (esketamine) marked a revolutionary departure from decades of reliance on serotonin-based therapies towards NMDAR modulation, representing the most significant breakthrough in psychiatry in decades. This paradigm shift is anchored by establishing esketamine as the gold standard option for TRD.10 Unlike traditional antidepressants that take weeks to alter mood, esketamine demonstrates that modulating the NMDAR can produce rapid, sustained antidepressant effects within hours in patients who have failed multiple previous therapies with selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs). This efficacy serves as proof-of-concept that the NMDAR is the critical control point for mood regulation. By triggering rapid synaptic repair (synaptogenesis) rather than just adjusting chemical imbalances, esketamine has proven that depression is a psychological disease with reversible structural deficits. Consequently, the pharmaceutical industry now views the NMDAR not just as an alternative target but as the central pillar for next-generation therapies, with the goal of replicating ketamine’s profound efficacy while overcoming its limitations.
The pharmaceutical industry now views the NMDAR not just as an alternative target but as the central pillar for next-generation therapies.
Current drug development is pivoting towards agents that can replicate esketamine’s rapid synaptic repair without its dissociative side effects. Strategies have largely focused on more selective molecules, such as glycine-site competitive modulators or specific GluN2B antagonists, attempting to refine the NMDAR’s status as a gatekeeper for emotional health. However, this approach has faced significant hurdles. Most notably, high-profile clinical trials for subunit-selective NMDAR antagonists, including Onfasprodil (Novartis), Traxoprodil (Pfizer) and EVT-101 (Evotec), have failed to demonstrate sufficient efficacy. These setbacks indicate that mere subunit-selective inhibition may not be potent enough to reproduce the robust anti-depressant effects of a non-selective antagonist like esketamine. Despite these challenges, the NMDAR remains the primary target for next-generation TRD therapies. The field is now witnessing intensive investigation into alternative strategies, ranging from the development of novel ‘ketamine mimics’ to the optimisation of oral ketamine formulations to overcome its limitations in oral bioavailability. These efforts aim to bridge the gap between efficacy and accessibility, striving to deliver the gold-standard benefits of NMDAR modulation in a convenient, patient-friendly format.
Breaking down the barriers for TRD treatment
However, current therapeutic strategies for TRD encounter a significant pharmacological hurdle often described as the ‘receptor occupancy trap’, which severely restricts the broader application of NMDAR antagonists. Although agents like ketamine and esketamine have transformed the field with their rapid antidepressant properties, their mechanism relies on physically blocking the NMDAR channel (Figure 1). To obtain the necessary therapeutic effect, these conventional antagonists must achieve high levels of target engagement/receptor occupancy – essentially inhibiting a vast proportion of the NMDA receptor pool in the brain to ensure the signal is dampened. The downside of this approach is that NMDARs are also indispensable for maintaining conscious awareness and sensory processing. Consequently, the high degree of blockage required to alleviate depression inevitably interferes with normal brain function, pushing the therapeutic dose to the threshold that triggers dissociation, hallucinations and sedation.
Current therapeutic strategies for TRD encounter a significant pharmacological hurdle often described as the ‘receptor occupancy trap’, which severely restricts the broader application of NMDAR antagonists.
These severe psychotomimetic side effects require high levels of risk management strategies; for instance, esketamine administration requires patients to be monitored by healthcare professionals for at least two hours after dosing. This requirement imposes a substantial logistical and financial burden on both patients and the healthcare system. Furthermore, this intrinsic link between efficacy and pharmacological side effects highlights a major challenge in current development efforts for conventional NMDAR antagonists, including reformulations and analogues of esketamine.
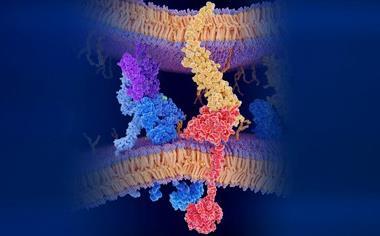
FundaMental Pharma is aiming to overcome this barrier by exploiting a fundamentally different and unique mechanism that combines the disruption of a specific protein–protein interaction with NMDAR channel blockade. Building on the seminal discovery of a novel class of compounds capable of disrupting the interaction between extrasynaptic NMDARs and the transient receptor potential cation channel subfamily M member 4 (TRPM4) ion channel,11 more potent molecules have been developed. These compounds uniquely combine this disruptive action with a conventional NMDAR channel blockade, thereby integrating two complementary mechanisms of action (Figure 1). Accordingly, FMP374, a clinical candidate for TRD, represents a novel class of molecules as dual-acting NMDAR modulators.
 Figure 1: Schematic image comparing the mechanism of action of conventional NMDAR antagonists with a novel dual-acting NMDAR modulator. Conventional NMDAR antagonists (eg, esketamine), which are mostly polycyclic aromatic compounds, act as open-channel blockers by entering the pore of the NMDAR channel. In contrast, FMP374 blocks the channel but also disrupts the NMDAR/TRPM4 protein complex.
Figure 1: Schematic image comparing the mechanism of action of conventional NMDAR antagonists with a novel dual-acting NMDAR modulator. Conventional NMDAR antagonists (eg, esketamine), which are mostly polycyclic aromatic compounds, act as open-channel blockers by entering the pore of the NMDAR channel. In contrast, FMP374 blocks the channel but also disrupts the NMDAR/TRPM4 protein complex.
[/caption]
FMP374 has shown remarkable preclinical efficacy and demonstrated rapid and sustained antidepressant effects in gold-standard depression models at low-nanomolar unbound CNS drug concentrations, with exceptional ≥10–20x margins over unwanted NMDAR-related side effects. Importantly, the candidate has shown no evidence of dissociation-like behaviours, hallucination-associated proxies, ataxia or hyperactivity at efficacious doses in various models, supporting its potential as an oral, at-home therapy for patients with TRD. Backed by robust preclinical data, FMP374 is ready for IND-enabling studies. Internal research suggests that FMP374 overcomes the receptor occupancy trap by requiring only low levels of NMDAR channel occupancy, likely due to the additional contribution of NMDAR/TRPM4 complex disruption to its preclinical efficacy. As expected for a modern antidepressant, FMP374 also acts as a potent neuroplastogen in vitro and in vivo, predictive of a long-lasting effect in TRD patients.
In conclusion, FMP374 offers a promising solution to the limitations of current therapies: a dual-acting NMDAR modulator capable of delivering potent, rapid relief from depression in an oral formulation that avoids the psychotomimetic effects that currently restrict the clinical use of NMDAR-targeting drugs.
References
- Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14, 383-400 (2013). https://doi.org:10.1038/nrn3504
- Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11, 682-696 (2010). https://doi.org:10.1038/nrn2911
- Lipton SA. Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx 1, 101-110 (2004). https://doi.org:10.1602/neurorx.1.1.101
- Reisberg B, et al. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 348, 1333-1341 (2003). https://doi.org:10.1056/NEJMoa013128
- Canuso CM, et al. Efficacy and Safety of Intranasal Esketamine for the Rapid Reduction of Symptoms of Depression and Suicidality in Patients at Imminent Risk for Suicide: Results of a Double-Blind, Randomized, Placebo-Controlled Study. Am J Psychiatry 175, 620-630 (2018). https://doi.org:10.1176/appi.ajp.2018.17060720
- Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 27, 11496-11500 (2007). https://doi.org:10.1523/JNEUROSCI.2213-07.2007
- Krystal JH, Abdallah CG, Sanacora G, et al. Ketamine: A Paradigm Shift for Depression Research and Treatment. Neuron 101, 774-778 (2019). https://doi.org:10.1016/j.neuron.2019.02.005
- McCarthy B, et al. Dextromethorphan-bupropion (Auvelity) for the Treatment of Major Depressive Disorder. Clin Psychopharmacol Neurosci 21, 609-616 (2023). https://doi.org:10.9758/cpn.23.1081
- Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 7, 426-437 (2008). https://doi.org:10.1038/nrd2462
- Moore TJ, Alami A, Alexander GC, Mattison DR. Safety and effectiveness of NMDA receptor antagonists for depression: A multidisciplinary review. Pharmacotherapy 42, 567-579 (2022). https://doi.org:10.1002/phar.2707
- Yan J, Bengtson CP, Buchthal B, et al. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 370 (2020). https://doi.org:10.1126/science.aay3302
About the authors
 Dirk Beher serves as Chief Executive Officer and executive member of the board of directors at FundaMental Pharma.
Dirk Beher serves as Chief Executive Officer and executive member of the board of directors at FundaMental Pharma.
He is an experienced biotech executive, serial CEO, board member, drug hunter and entrepreneur. In addition to his executive background, his long-standing (>25 years) CNS drug discovery and development experience is particularly relevant to FundaMental Pharma’s ambition to overcome barriers in treatment-resistant depression. He previously co-founded and served for a decade as the CEO of Asceneuron.
Prior to biotech, Dirk spent 14 years in biopharmaceutical drug discovery leading CNS-focused R&D teams around the globe at Merck Sharp & Dohme (Merck & Co.; UK), Amgen (US) and Merck Serono (CH).
Dirk holds a German Diploma (MSc) and PhD from the Heidelberg University, Germany. He has authored over 50 peer reviewed scientific articles & reviews and is an inventor of multiple granted and pending patents.
 Jing Yan serves as senior Director of Research at FundaMental Pharma.
Jing Yan serves as senior Director of Research at FundaMental Pharma.
He is an experienced neuroscientist and pioneered the discovery of our dual-acting NMDA receptor modulators and their role in depression.
His extensive experience in CNS research and drug development directly supports FundaMental Pharma’s mission to overcome barriers in treatment-resistant depression.
Jing Yan is a neuroscientist with over a decade of experience in CNS research, specialising in neurological diseases. His breakthrough discovery of the NMDAR/TRPM4 complex was published in Science and laid the scientific foundation for new therapeutic approaches.
His research has been distinguished with some of most prestigious scientific honors, including the Karl-Freudenberg-Preis of the Heidelberg Academy of Sciences, the Ruprecht-Karls-Preis of Heidelberg University and the IZN/Chica and Heinz Schaller Young Investigator Neuroscience Award.
Jing Yan earned his PhD in Neurobiology (summa cum laude) from Heidelberg University (Germany). He has published as first author in leading journals including Science, Cell Reports Medicine and Molecular Psychiatry and is an inventor on multiple granted and pending patents in the field of neurological diseases.