



By combining CRISPR knock-in with small peptide tags, researchers can study proteins in their native cellular context, generating more predictive data for translational drug discovery.
Drug discovery is moving towards more complex intracellular targets – from transcription factors and chromatin remodellers to intrinsically disordered proteins – that have proved challenging to study using conventional methods. Overexpression systems flood the cell with non-physiological levels of protein; antibodies vary in availability, specificity and reproducibility; and many of the most therapeutically interesting proteins exist at low abundance under tight regulatory control. The result is a translational gap where in vitro and biochemical readouts that look promising at the screening stage often fail to predict behaviour in disease-relevant cellular contexts. Fortunately, an alternative approach has been maturing over the past decade: studying proteins endogenously with genome editing.
Why small tags matter: preserving native protein function
The biological case for endogenous tagging comes down to one principle: a protein is not just its sequence – it is its sequence combined with expression at the right level, in the right place and under the right regulatory controls. Tagging the protein at its native locus preserves all of these, while introducing the gene from outside the cell does not. Overexpression from a strong constitutive promoter routinely expresses protein at orders of magnitude above physiological levels; this can saturate degradation machinery, force mislocalisation and obscure the feedback loops that normally constrain protein abundance. The result can be compensatory responses from the cell, stoichiometric imbalances with binding partners or aberrant post-translational modifications.1
The biological case for endogenous tagging comes down to one principle: a protein is not just its sequence – it is its sequence combined with expression at the right level, in the right place and under the right regulatory controls.
Knock-in tagging at the endogenous locus avoids those pitfalls, but only if the tag itself does not introduce new ones. This is where size matters. Larger fusion partners – full-length fluorescent proteins such as GFP or mCherry are around 25–30kDa – can disrupt folding, alter trafficking, mask interaction surfaces or change turnover kinetics, particularly for small or structurally constrained targets. Small peptide tags, by contrast, are short enough to be placed at either terminus, or even at internal flexible loops, with minimal effect on protein function.2
Several small epitope tags have been the workhorses of this approach for decades. FLAG (eight amino acids), HA (nine amino acids derived from influenza haemagglutinin), Myc (10 amino acids) and V5 (14 amino acids derived from simian virus 5) are all under 1.5kDa and are detected through well-characterised monoclonal antibodies.2 Originally developed for use in overexpression constructs, these tags have proven equally well suited to endogenous knock-in, where their small footprint reduces the risk that the tagging itself confounds the experiment. Their long history of use also means that suitable detection reagents and protocols are widely standardised. Newer-generation tags, such as HiBiT, have added another option to the small-tag toolbox. At 11 amino acids, HiBiT is comparable in size to FLAG, HA or Myc and can be inserted at endogenous loci in the same way.3 What distinguishes it is detection flexibility: HiBiT supports both highly sensitive bioluminescent readouts through split complementation, powered by NanoLuc luciferase, and conventional antibody-based detection.
The common thread across all these tools is that they are small enough not to perturb protein physiology, while providing a protein-agnostic method of measurement. Importantly, small peptide tagging generalises across very different classes of target, including proteins that span a wide range of sizes, subcellular localisations, expression levels and functions.4 Taken together, the case for tagging endogenous proteins with short peptides in translational models is strong on biological grounds. Whether that case translates into routine practice across discovery pipelines, however, depends on how reliably and quickly the underlying cell lines can be generated in the first place.
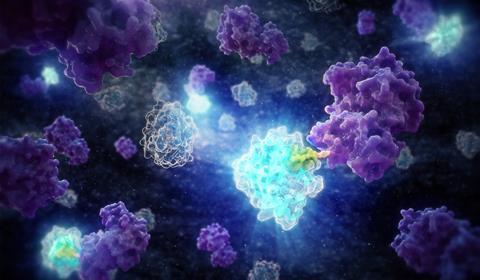
By inserting a small peptide tag such as HiBiT (green) directly into the endogenous locus, proteins (purple) can be detected at physiological levels through bioluminescent complementation with LgBiT (blue), without the artefacts introduced by overexpression or bulky fluorescent fusions. Credit: Promega UK
If endogenous tagging is so attractive, why is it not the default? The answer, historically, has been efficiency. CRISPR/Cas9 readily generates a double-strand break, but the homology-directed repair (HDR) pathway required for precise sequence insertion is slow, cell-cycle restricted and outcompeted by non-homologous end joining (NHEJ) in most cell types.5,6 Reported HDR rates in primary cells, induced pluripotent stem cells (iPSCs) and many cancer lines have often sat in the low single digits, making the generation of correctly edited clones laborious and unpredictable.
If endogenous tagging is so attractive, why is it not the default? The answer, historically, has been efficiency.
Several technical advances have shifted that landscape. Donor template design has been a major focus, with linearised donors, single-stranded DNA donors carrying optimised homology arms and asymmetric homology designs all raising insertion frequency substantially. Combined with better guide RNA selection, ribonucleoprotein delivery of Cas9 and modulation of repair pathway choice, these advances have made knock-in editing routine in cell types that were once considered intractable.6-8
With those gains has come scalability. The time and cost of moving from designing a knock-in to generating data have fallen sharply as bioinformatics and laboratory automation have been integrated into routine workflows, with the added benefit of greater consistency between batches of edited cells. End-to-end packages covering design, editing, sequencing-based validation of on-target integration and zygosity confirmation are now widely available – and the two routes by which researchers access them tell their own story about how the technology has matured.
On the commercial side, providers such as California-based EditCo have invested in highly parallelised automated platforms, making it economically practical to commission editing campaigns across many targets at once rather than treating each as a bespoke project. On the academic side, dedicated core facilities have taken up the same workflows. The Genome Editing Unit at the University of Manchester, for example, applies CRISPR routinely to translational research models including reporter-tagged cell lines, pluripotent stem cells and fruit flies. The fact that the same technology underpins commercial and academic infrastructure alike signals where the field is heading.
Applications in discovery pipelines
Targeted protein degradation has become one of the most visible drivers of demand for endogenous tagging. PROTACs, molecular glues and related modalities act by inducing degradation rather than inhibiting activity, so the relevant pharmacological readout is a quantitative change in endogenous protein level over time. A tagged cell line provides that readout directly, with the choice of tag determining how the measurement is made: immunoblot quantification measures total protein levels at defined time points, while tags compatible with live-cell readouts allow degradation kinetics to be sampled continuously.9 Both approaches yield data on maximal degradation, recovery and dose-response, but they depend on the protein being expressed at native levels rather than from a transgene that can saturate or escape the degradation machinery being studied.
Targeted protein degradation has become one of the most visible drivers of demand for endogenous tagging.
Target validation is a second area where the approach pays off. Confirming that a candidate target behaves as hypothesised – eg, that it is expressed in the disease-relevant cell type and its abundance correlates with the phenotype of interest – is far more credible when measurements come from the endogenous protein. Tagged proteins also simplify protein–protein interaction studies that can be supported by co-immunoprecipitation from endogenous lysates.
Live-cell functional assays form the final pillar. Subcellular trafficking, response to receptor activation, induction by transcriptional stimuli and turnover under stress can all be followed in tagged cells using whichever detection modality best suits the question. Spatial information is accessible through immunofluorescence or bioluminescence imaging, while quantitative kinetics are best captured by plate-based bioluminescence readouts. For dynamic, signalling-driven processes that dominate the current pipeline of challenging targets, observing protein behaviour across minutes to hours in intact cells is qualitatively different from the static snapshots offered at discrete timepoints.
The future of endogenous biology
The combination of CRISPR knock-in and small-peptide tagging is not a single product or workflow, but a methodological shift in how endogenous proteins are studied. Established epitope tags such as FLAG, HA, Myc and V5 continue to provide a reliable, antibody-based foundation, while newer entries such as HiBiT extend the toolkit by enabling sensitive bioluminescent detection alongside conventional antibody readouts from the same insertion. As editing efficiency continues to improve and as more cell models, including from iPSC-derived and patient-derived backgrounds, become accessible to robust knock-in protocols, the proportion of discovery work performed on endogenously tagged systems will keep growing. The implication for translation is straightforward: assays run on proteins expressed at native levels in cells of relevant biology are more likely to produce data that will be predictive in more complex models and ultimately the clinic. For drug discovery teams confronting intracellular targets, dynamic protein systems and the demands of new modalities such as targeted degradation, endogenous tagging is fast becoming indispensable for modern drug discovery.
References
1. Gibson TJ, Seiler M, Veitia RA. The transience of transient overexpression. Nat Methods. 2013;10(8):715-721.
2. Brizzard B. Epitope tagging. Biotechniques. 2008;44(5):693-695.
3. Schwinn MK, Machleidt T, Zimmerman K, et al. CRISPR-mediated tagging of endogenous proteins with a luminescent peptide. ACS Chem Biol. 2018;13(2):467-474.
4. Schwinn MK, Steffen LS, Zimmerman K, et al. A simple and scalable strategy for analysis of endogenous protein dynamics. Sci Rep. 2020;10:8953.
5. Liu M, Rehman S, Tang X, et al. Methodologies for improving HDR efficiency. Front Genet. 2019;9:691.
6. Zhang JP, Li XL, Li GH, et al. Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol. 2017;18(1):35.
7. Richardson CD, Ray GJ, DeWitt MA, et al. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34(3):339-344.
8. Riesenberg S, Maricic T. Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Nat Commun. 2018;9(1):2164.
9. Riching KM, Mahan S, Corona CR, et al. Quantitative live-cell kinetic degradation and mechanistic profiling of PROTAC mode of action. ACS Chem Biol. 2018;13(9):2758-2770.












No comments yet