



Dr Aaron Wenger reveals how improvements in long-read sequencing technology is enabling the elucidation of complex disease mechanisms for targeted and effective treatments for rare diseases.

Drug discovery seeks to target clear biological mechanisms. Yet in rare disease research, many of the most consequential mechanisms sit within complex regions of the genome that have historically been difficult to analyse. These hidden regions often underpin severe, inherited conditions, but when the underlying variation cannot be fully resolved, targets emerging from these regions carry too much ambiguity to support high-risk development programmes.
Fortunately that constraint is beginning to lift, as demonstrated by a milestone therapeutic development in 2025 for Huntington’s disease. A new therapy demonstrated sustained slowing of progression in the disease, which was long considered a difficult target due to underlying repeat expansion, illustrating what is possible when complex genomic architecture is defined with precision. Greater clarity at the sequence level enables new therapeutic strategies to be designed, strengthening the link between genetic insight and clinical intervention. Regions once regarded as technically out of reach are now entering the arena of viable target discovery, reshaping how rare disease biology is translated into precision therapeutics.
Sequencing constraints and the complexity gap
One reason scientists struggle to understand complex regions associated with rare disease is the reliance on short-read sequencing. This approach fragments DNA into small pieces, sequences them individually, then aligns the reads back to a reference genome. While powerful and scalable, the short read length makes it difficult to reconstruct long repetitive stretches, disentangle highly similar gene copies or fully characterise structural variation. The result is gaps in interpretation and uncertainty in variant calling. For rare disease research, this means that genuine pathogenic mechanisms are sometimes only partially characterised or else misclassified entirely.
In contrast, long-read sequencing captures extended DNA molecules in single contiguous reads. Entire repeat regions can be sequenced without assembly, allowing precise sizing and structural characterisation. Long-read sequencing was once considered costly and limited in throughput. However, newer generations of long-read platforms deliver higher output at substantially lower cost, making a higher level of genomic resolution more accessible to research and translational settings.
As resolution improves, three categories of genomic complexity that have long hindered rare disease diagnostics and drug development are becoming tractable: repeat expansions, paralogous genes and epigenetic signals.
1. Repeat expansions
Repeat expansions are a class of mutation in which short DNA sequences are repeated multiple times in succession. In healthy individuals, these repeats fall within a stable range. When the number of repeats exceeds a pathogenic threshold, gene function can be disrupted, leading to progressive neurological and developmental disorders.
Huntington’s disease is one of the most well-known examples of this, caused by expansion of a CAG trinucleotide repeat within the HTT gene. When this repeat grows beyond a defined limit, toxic protein aggregates form and drive neurodegeneration.
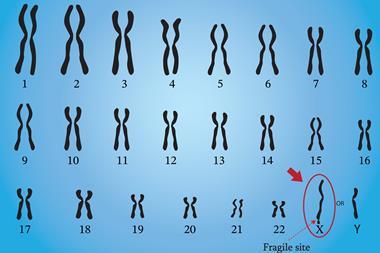
Huntington’s disease is one of the most well-known examples of this, caused by expansion of a CAG trinucleotide repeat within the HTT gene. When this repeat grows beyond a defined limit, toxic protein aggregates form and drive neurodegeneration. Other conditions – including certain forms of amyotrophic lateral sclerosis (ALS), frontotemporal dementia and fragile X syndrome – are also driven by pathogenic repeat expansions.
Short-read sequencing struggles with repeat expansions because fragmented reads cannot reliably span long, repetitive stretches. The true length and full sequence of the repeat are obscured, complicating variant interpretation and introducing uncertainty. The clarity delivered by long-read sequencing strengthens confidence in the causal mechanism and supports the development of targeted interventions, including antisense oligonucleotide therapies designed to modulate repeat-driven transcripts.
2. Paralogous genes
Paralogous genes are duplicated copies of a gene that share very similar DNA sequences. Many sit within segmental duplications: large, repeated regions of the genome that are difficult to analyse. Although these regions contain medically important genes, their similarity has historically made it hard to distinguish one copy from another with confidence.
Spinal muscular atrophy (SMA) illustrates the challenge. This condition is caused by changes in the SMN1 gene, but a nearly identical copy, SMN2, sits alongside it. The number and integrity of SMN2 copies influence disease severity and treatment response. Accurately identifying which gene copy carries a variant and how many copies are present is essential for both diagnosis and therapeutic decision making.
Short-read sequencing often cannot reliably assign variants to the correct paralogue because fragmented reads lack sufficient context.
Short-read sequencing often cannot reliably assign variants to the correct paralogue because fragmented reads lack sufficient context. This introduces uncertainty in variant calling and copy number estimation, increasing risk in both variant interpretation and target validation.
Long-read sequencing provides extended DNA reads that span distinguishing regions, enabling accurate phasing and copy number assessment within duplicated loci. For drug discovery, that clarity turns paralogous regions from technical obstacles into defined and potentially viable therapeutic targets.
3. Epigenetic signals
Not all rare diseases are driven solely by changes in the DNA sequence itself. In some cases, the underlying code is intact, but the way genes are regulated is altered. Epigenetic modifications, such as DNA methylation, can switch genes on or off without changing the DNA sequence. When these regulatory signals go awry, the consequences can be as severe as a mutation.
Epigenetic alterations are increasingly recognised in neurodevelopmental and metabolic disorders. Traditional sequencing approaches focus primarily on identifying sequence variants and often require separate assays to detect epigenetic changes. This fragmented view can leave important regulatory mechanisms unexplored.
Traditional sequencing approaches focus primarily on identifying sequence variants and often require separate assays to detect epigenetic changes. This fragmented view can leave important regulatory mechanisms unexplored.
Long-read sequencing enables the detection of methylation patterns directly from native DNA alongside sequence information. This integrated view allows researchers to see both the genetic variant and its regulatory context in a single experiment. For target discovery, this added layer of insight is critical. By capturing epigenetic signals together with sequence data, researchers move closer to defining targets based on function rather than sequence alone.
A compelling example of this comes from a University of Washington study in which researchers sequenced the genome, transcriptome, methylome and chromatin epigenome simultaneously in an undiagnosed nine-month-old female with multiple phenotypic abnormalities. While genomic analysis identified NBEA haploinsufficiency as the cause of the child’s developmental delay, it was multiomic profiling that revealed a distinct epigenetic driver for her bilateral retinoblastomas. Because this alteration did not change the underlying DNA sequence, it would have been missed by genome sequencing alone. The study illustrates how integrating epigenetic data alongside sequence information can uncover clinically relevant mechanisms invisible to single-omic approaches.
Redefining viable targets
In rare disease research, where patient numbers are small and development risk is high, precision is a prerequisite. Clearer genomic definition reduces uncertainty at the point of target selection, enabling informed decisions about which programmes warrant investment and which should be deprioritised. Greater resolution also creates opportunities for therapeutic repurposing. When complex variants are characterised with accuracy, existing drugs or platform technologies can be assessed against newly defined mechanisms, shortening the path from insight to intervention.
As tools to resolve genomic complexity continue to mature, the scope of what constitutes a viable target will broaden, opening new avenues for precision therapeutics for rare diseases built on biological certainty rather than approximation.










No comments yet