

Using clinical drug resistance to kill cancer cells
Posted: 11 July 2017 | Howard Fearnhead - NUI Galway, Sheraz Gul (Fraunhofer-IME SP) | No comments yet
Drug resistance is a significant therapeutic problem, from infectious diseases to cancer. Given a large enough population of genetically varying target cells, the emergence of drug resistance is not surprising and is even unavoidable…

What is worse is that cells can become resistant to multiple drugs with different mechanisms of action because drugs induce shared stress and cell-death pathways. Despite this dire situation, drug-resistant cells are not invulnerable. New results suggest that ‘collateral sensitivity cycling’, in which drugs are used sequentially to select against drug resistance, may be generally applicable to infectious diseases and cancer.
Collateral sensitivity describes a situation in which a cell becomes resistant to a drug, but the very mechanism of resistance to that drug brings an increased vulnerability to a second, different drug. Collateral sensitivity has been observed in bacteria and offers a way to attack the growing problem of antibiotic resistance.1 Although the emergence of new molecular tools means the idea is undergoing a period of renewed interest, it is an old concept, with one of the earliest uses of the term being in 1958.2
In a more recent study3 to identify examples of collateral sensitivity, E.coli were treated with antibiotics to establish approximately 20 different resistant strains. Each strain was then tested for its sensitivity to other antibiotics. This approach identified pairs of antibiotics in which resistance to one drug enhanced sensitivity to the other. More complex relationships involving three drugs were also identified such that resistance to drug ‘A’ caused sensitivity to ‘B’, and resistance to ‘B’ caused sensitivity to ‘C’.
These relationships suggest appropriate cyclical treatment with specific antibiotics could be a valuable strategy when facing drug resistance that does not require the discovery of new antibacterial drugs. Importantly, the proper use of antibiotics in this way will slow the appearance of multi-drug resistant bacteria4 as well as offering viable strategies for significant clinical problems of drug resistance.5
Collateral sensitivity and anti-cancer agents
In vitro studies of cancer cells support the idea that collateral sensitivity could be exploited to kill drug-resistant tumours.6 The MDR1 gene encodes an ATP-dependent pump that extrudes cytotoxic drugs from cells and is upregulated in some tumours. MDR1 overexpressing cells are more sensitive to some anti-cancer nucleotide analogues and to small molecules identified by screening. The orphan drug tiopronin can kill cancer cells expressing high levels of MDR1.7,8 Tiopronin acts by inhibiting glutathione peroxidase, although why MDR1 overexpressing cells are more sensitive is unclear, and the clinical usefulness of tiopronin has not been tested. It is likely that the principle of collateral sensitivity can be exploited to address other clinically important forms of drug resistance. A recent study reported collateral sensitivity in a murine model of acute lymphoblastic leukaemia treated with BCR-ABL inhibitors.9
Drug resistance in colorectal cancer
Colorectal cancer is the third biggest killer of women, and the second of men (www.cancer.org). 5-fluorouracil (5-FU) is the main pharmacological treatment for colorectal cancer, and while many initially respond well to 5-FU, recurrence of the tumour is typically accompanied by resistance to 5-FU. In addition, the metastases in those with advanced disease are also resistant to 5-FU (Figure 1). The major molecular mechanism of 5-FU resistance in patients with colorectal cancer is known, and it can be modelled using in vitro systems. This combination of a large unmet medical need with a molecular understanding and relevant experimental models provides a strong rationale for testing whether collateral sensitivity can be exploited to kill 5-FU resistant cancers.
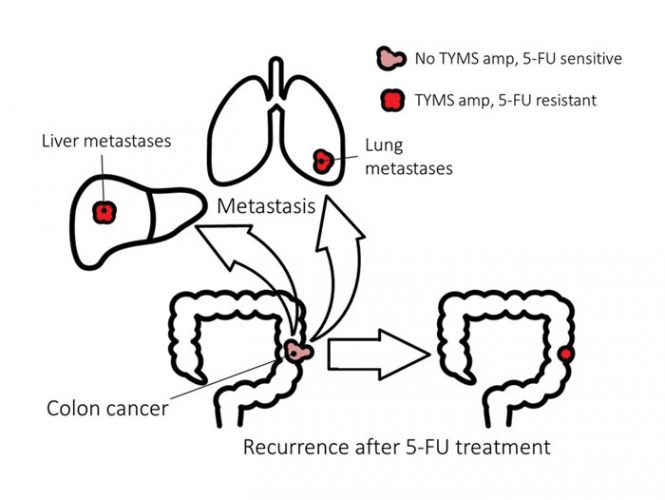
Figure 1.
5-FU exerts its anti-cancer activity primarily through inhibition of thymidylate synthetase (TYMS). TYMS catalyses the methylation of deoxyuridine monophosphate (dUMP) to form thymidylate (dTMP) using 5,10-methylenetetrahydrofolate as a methyl donor. dTMP is then phosphorylated to produce deoxythymidine triphosphate (dTTP) for DNA replication and repair. Inhibition of TYMS depletes the cell of dTTP, perturbing the levels of the other deoxynucleotides, compromising DNA synthesis and repair and causing lethal DNA damage. 5-FU can also be misincorporated into RNA and DNA and both of these mechanisms may also contribute to cancer cell death. 5-FU can affect RNA in various ways, including inhibiting the maturation of pre-rRNA, disrupting post-transcriptional modification of RNAs, and inhibiting splicing of pre-mRNA.
Drug activation
5-FU is a prodrug that undergoes metabolic activation; thymidine phosphorylase (TP) converts 5-FU to fluorodeoxyuridine, which is converted to the active metabolite fluorodeoxyuridine monophosphate (FdUMP). TP overexpression in cell culture and xenograft models increases the sensitivity of cancer cells to 5-FU,10,11 but there is no evidence that colorectal tumours with high TP levels respond well to 5-FU.12
Drug inactivation
5-FU can also undergo metabolism by dihydropyrimidine dehydrogenase (DPD) to an inactive form (5-FUH2), reducing the concentration of 5-FU and in turn limiting the concentration of FdUMP. DPD polymorphisms are associated with an increased risk of severe dose-limiting toxicities.13,14 At the same time, in vitro overexpression of DPD in cancer cell lines confers resistance to 5-FU15 and high DPD mRNA levels in colorectal tumours correlates with resistance to 5-FU.16 EGFR mutations in lung cancer patients also correlate with increased levels of DPD and a poor response to 5-FU.17 These increased DPD levels may shield the tumour from 5-FU by rapidly inactivating the drug.
TYMS translation and mutation
TYMS binds to and regulates the translation of its own mRNA. This regulation is disrupted by 5-FU such that TYMS protein bound to FdUMP cannot inhibit the translation of TYMS mRNA. Thus 5-FU treatment increases TYMS protein, decreasing sensitivity to 5-FU.
Overexpression of thymidine kinase, which phosphorylates thymidine to make dTMP, can provide dTMP in the absence of TYMS activity and can confer resistance to 5-FU.18 TYMS point mutants (D49G and G52S) were identified in HT1080 human sarcoma cells after ethyl methanesulphonate mutagenesis and selection with an anti-folate.19 These mutants retain enzymatic activity but cause resistance to 5-FU, possibly by perturbing the structure of a highly conserved Arg50 loop. A Y33H substitution can also increase resistance to 5-FU,20 but link between TYMS point mutations and drug-resistant tumours is weak.21
Increased levels of TYMS
Overexpression of TYMS in cancer cell lines increases the resistance of the cells to 5-FU.22 A similar picture emerged from experiments using a TYMS-inducible cancer cell line.23 In-vitro exposure of cancer cell lines to 5-FU can select for cells with amplification of the TYMS locus leading to elevated TYMS mRNA and protein, and an increased resistance to 5-FU.24-27 In addition, amplification of the TYMS locus is detected in patients with advanced, metastatic colorectal cancer and who had previously received 5-FU treatment.28-30 The increase in copy number of TYMS correlated with poorer survival.
Epigenetics and 5-FU treatment
Colorectal cancer lines selected for resistance to 5-FU by long-term 5-FU treatment have been used to investigate whether epigenetic factors contribute to drug resistance. These studies have shown that DNA methyltransferase inhibitors can increase the 5-FU sensitivity of 5-FU resistant colorectal cancer cells. However, the mechanism of 5-FU resistance was not established in these studies and so the link between TYMS amplification and epigenetic regulation of 5-FU sensitivity is not known.
Clinically relevant mechanisms
Thus, several different resistance mechanisms have been identified in cancer research models, but TYMS amplification emerges as the mechanism most strongly associated with clinical resistance and metastases. While TYMS amplification is currently an indicator of poor prognosis, the examples of collateral sensitivity from bacterial disease and in other cancer situations suggests that a rational response to this problem could be developed.
Finding new treatments
The strategy is simple in principle. Use a 5-FU sensitive cell line and a second cell line that is derived from the first, but has TYMS amplification and is 5-FU resistant, to identify agents that are selectively toxic to the second cell line. There are different ways to obtain these pairs, both with their own advantages and disadvantages.
One approach is to select in vitro for 5-FU resistant colorectal cancer cells using increasing concentrations of 5-FU over a period of several months. This approach resembles the in vivo situation in the sense that the drug itself is used to make resistant cells and the approach has been used to make resistant cancer lines with TYMS amplification. However, as other molecular mechanisms of resistance exist, and the selection takes place in vitro without a proper tumour context, the resistant cells need not have TYMS amplification or be a clinically relevant model. If a chemical agent does show increased activity against 5-FU resistant cells made by selection, RNAi would be needed to test whether increased TYMS levels cause the altered sensitivity.
An alternative is to overexpress the TYMS gene under the control of a strong promoter. This provides a model system that can be used to robustly test whether TYMS levels increase the sensitivity of a cell to a chemical agent. By mimicking amplification, it also resembles the clinical situation. But not completely: the most commonly amplified locus contains TYMS and a second gene, rTS. Alternative splicing allows rTS to encode an antisense transcript to the terminal intron and 3’-UTR of TYMS and two proteins, rTSβ and rTSg. The antisense transcript destabilises TMYS mRNA and so downregulates TYMS.31 rTSβ encodes an enzyme that also regulates TMYS, possibly through the generation of a small signalling molecule. Both the anti-sense transcript and rTSβ may cause wider changes in the cell by affecting processes beyond TYMS regulation. The transcript may act as a microRNA sponge, perturbing microRNA signalling and small signalling molecules may target other cellular pathways. Ideally an overexpression model should reflect this complexity.
 We are currently approaching this by using 5-FU sensitive colorectal cancer cell lines expressing red fluorescent protein and green fluorescent protein expressing 5-FU resistant cells (which also overexpress TYMS). Chemical libraries can be screened using a mixture of the two populations and cell death is assessed using a Perkin Elmer Operetta system after staining with vital dyes. This allows simultaneous assessment of both sensitive and resistant cells.
We are currently approaching this by using 5-FU sensitive colorectal cancer cell lines expressing red fluorescent protein and green fluorescent protein expressing 5-FU resistant cells (which also overexpress TYMS). Chemical libraries can be screened using a mixture of the two populations and cell death is assessed using a Perkin Elmer Operetta system after staining with vital dyes. This allows simultaneous assessment of both sensitive and resistant cells.
Candidate-based approaches
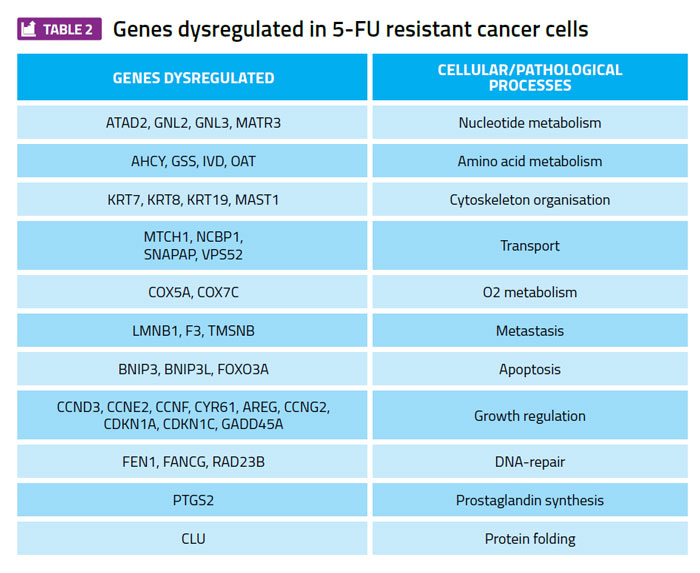
Profiling gene expression patterns in cultured cancer cell lines and searching for differences between 5-FU sensitive and 5-FU resistant cell lines has identified many examples of genes whose expression level correlates with 5-FU sensitivity (Table 2). These genes are candidates for a focused RNAi approach to test whether any of these genes are required to maintain the viability of TYMS overexpressing cells, but are not required for the viability of the 5-FU sensitive parental cell lines. A gene that meets these criteria is a potential therapeutic target for further investigation and development.
What to screen?
Following the example from the world of bacteria and antibiotics, the first step should be to screen clinically approved compounds to identify existing drugs that might be repurposed to treat advanced colorectal cancer. If this strategy fails, screening of small molecule libraries covering drug-like chemical space or natural product libraries should be used to assess the utility of the collateral sensitivity approach as quickly as possible.
Author biographies
 HOWARD FEARNHEAD trained in pharmacology and toxicology at the London School of Pharmacy, and the MRC-Toxicology Centre, Leicester, UK. After a post-doctoral fellowship in the USA, he led a research team at the National Cancer Institute, NIH, before moving to the National University of Ireland, Galway. He researches cell death mechanisms with the goal of understanding how cancer cells make life-and-death decisions.
HOWARD FEARNHEAD trained in pharmacology and toxicology at the London School of Pharmacy, and the MRC-Toxicology Centre, Leicester, UK. After a post-doctoral fellowship in the USA, he led a research team at the National Cancer Institute, NIH, before moving to the National University of Ireland, Galway. He researches cell death mechanisms with the goal of understanding how cancer cells make life-and-death decisions.
 SHERAZ GUL is the Head of Assay Development and Screening at the Fraunhofer- IME SP, Hamburg. He has 23 years’ experience in both academia (University of London) and industry (GlaxoSmithKline). This has ranged from the detailed study of biological catalysts to the design and development of assays for high-throughput screening for the major drug target classes.
SHERAZ GUL is the Head of Assay Development and Screening at the Fraunhofer- IME SP, Hamburg. He has 23 years’ experience in both academia (University of London) and industry (GlaxoSmithKline). This has ranged from the detailed study of biological catalysts to the design and development of assays for high-throughput screening for the major drug target classes.
References
- Pal C, Papp B, Lazar V. Collateral sensitivity of antibiotic-resistant microbes. Trends Microbiol. 2015;23:401-7.
- Guthrie R, Loebeck ME, Hillman MJ, Zgorzynski F. Tests for collateral sensitivity or cross resistance in bacterial mutants resistant to amethopterin or purine analogs. Cancer Res. 1958;18:319-34.
- Imamovic L, Sommer MO. Use of collateral sensitivity networks to design drug cycling protocols that avoid resistance development. Sci Transl Med. 2013;5: 204ra132.
- Kim S, Lieberman TD, Kishony R. Alternating antibiotic treatments constrain evolutionary paths to multidrug resistance. Proc Natl Acad Sci USA. 2014;111: 14494-9.
- Gonzales PR, Pesesky MW, Bouley R, Ballard A, Biddy BA, Suckow MA, Wolter WR, Schroeder VA, Burnham CA, Mobashery S, Chang M, Dantas G. 2015. Synergistic, collaterally sensitive beta-lactam combinations suppress resistance in MRSA. Nat Chem Biol. 2015;11:855-61.
- Pluchino KM, Hall MD, Goldsborough AS, Callaghan R, Gottesman MM. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist Updat. 2012; 15:98-105.
- Goldsborough AS, Handley MD, Dulcey AE, Pluchino KM, Kannan P, Brimacombe KR, Hall MD, Griffiths G, Gottesman MM. Collateral sensitivity of multidrug-resistant cells to the orphan drug tiopronin. J Med Chem. 2011;54:4987-97.
- Hall MD, Marshall TS, Kwit AD, Miller Jenkins LM, Dulcey AE, Madigan JP, Pluchino KM, Goldsborough AS, Brimacombe KR, Griffiths GL, Gottesman MM. Inhibition of glutathione peroxidase mediates the collateral sensitivity of multidrug-resistant cells to tiopronin. J Biol Chem. 2014;289:21473-89.
- Zhao B, Sedlak JC, Srinivas R, Creixell P, Pritchard JR, Tidor B, Lauffenburger DA, Hemann MT. Exploiting Temporal Collateral Sensitivity in Tumor Clonal Evolution. Cell. 2016;165:234-46.
- Evrard A, Cuq P, Ciccolini J, Vian, l. & cano, j. p. 1999. Increased cytotoxicity and bystander effect of 5-fluorouracil and 5-deoxy-5-fluorouridine in human colorectal cancer cells transfected with thymidine phosphorylase. Br J Cancer, 80, 1726-33.
- Evrard A, Cuq P Robert B, Vian L, Pelegrin A, Cano JP. 1999. Enhancement of 5-fluorouracil cytotoxicity by human thymidine-phosphorylase expression in cancer cells: in vitro and in vivo study. Int J Cancer. 1999;80:465-70.
- Metzger R, Danenberg K, Leichman CG, Salonga D, Schwartz EL, Wadler S, Lenz HJ, Groshen S, Leichman L, Danenberg PV. 1998. High basal level gene expression of thymidine phosphorylase (platelet-derived endothelial cell growth factor) in colorectal tumors is associated with nonresponse to 5-fluorouracil. Clin Cancer Res, 4, 2371-6.
- Johnson MR, Hageboutros A, Wang K, High L, Smith JB, Diasio RB. 1999. Life-threatening toxicity in a dihydropyrimidine dehydrogenase-deficient patient after treatment with topical 5-fluorouracil. Clin Cancer Res. 1999;5:2006-11.
- Baskin Y, Amirfallah A, Unal OU, Calibasi G, Oztop I. Dihydropyrimidine dehydrogenase 85T>C mutation is associated with ocular toxicity of 5-fluorouracil: a case report. Am J Ther. 2015;22:e36-9.
- Takebe N, Zhao SC, Ural AU, Johnson MR, Banerjee D, Diasio RB, Bertino JR. Retroviral transduction of human dihydropyrimidine dehydrogenase cDNA confers resistance to 5-fluorouracil in murine hematopoietic progenitor cells and human CD34+-enriched peripheral blood progenitor cells. Cancer Gene Ther. 2001;8:966-73.
- Salonga D, Danenberg KD, Johnson M, Metzger R, Groshen S, Tsao-wei, DD, Lenz HJ, Leichman CG, leichman L, Diasio RB, Danenberg PV. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res. 2000;6:1322-7.
- Mochinaga K, Tsuchiya T, Nagasaki T, Arai J, Tominaga T, Yamasaki N, Matsumoto K, Miyazaki T, Nanashima A, Hayashi T, Tsukamoto K, Nagayasu T. High expression of dihydropyrimidine dehydrogenase in lung adenocarcinoma is associated with mutations in epidermal growth factor receptor: implications for the treatment of non–small-cell lung cancer using 5-fluorouracil. Clin Lung Cancer. 2014;15:136-144 e4.
- Fanciullino R, Evrard A, Cuq P, Giacometti S, Peillard L, Mercier C, Aubert C, Milano G, Ciccolini J. Genetic and biochemical modulation of 5-fluorouracil through the overexpression of thymidine kinase: an in-vitro study. Anticancer Drugs. 2006;17, 463-70.
- Tong Y, Liu-Chen X, Ercikan-Abali EA, Capiaux GM, Zhao SC, Banerjee D, Bertino, JR. Isolation and characterization of thymitaq (AG337) and 5-fluoro-2-deoxyuridylate-resistant mutants of human thymidylate synthase from ethyl methanesulfonate-exposed human sarcoma HT1080 cells. J Biol Chem. 1998;273: 11611-8.
- Hughey CT, Barbour KW, Berger FG, Berger SH. Functional effects of a naturally occurring amino acid substitution in human thymidylate synthase. Mol Pharmacol. 1993;44:316-23.
- Sanguedolce R, Alessandro R, De Leo G, Gullotti L, Sanguedolce F, Vultaggio G, Diana G, Cirello B, Rausa L. Failure of detection of the tyrosine to histidine substitution at the residue 33 of thymidylate synthase in human colorectal cancer. A preliminary study. Anticancer Res. 2000;20:4347-50.
- Saga Y, Suzuki M, Mizukami H, Kohno T, Takei Y, Fukushima M, Ozawa K. Overexpression of thymidylate synthase mediates desensitization for 5-fluorouracil of tumor cells. Int J Cancer. 2003;106:324-6.
- Longley DB, Ferguson PR, Boyer J, Latif T, Lynch M, Maxwell P, Harkin DP, Johnston PG. Characterization of a thymidylate synthase (TS)-inducible cell line: a model system for studying sensitivity to TS- and non-TS-targeted chemotherapies. Clin Cancer Res. 2001;7:3533-9.
- Copur S, Aiba K, Drake JC, Allegra CJ, Chu E. Thymidylate synthase gene amplification in human colon cancer cell lines resistant to 5-fluorouracil. Biochem Pharmacol. 1995;49:1419-26.
- Clark JL, Berger SH, Mittelman A, Berger FG. Thymidylate synthase gene amplification in a colon tumor resistant to fluoropyrimidine chemotherapy. Cancer Treat Rep. 1987;71:261-5.
- Berger SH, Jenh CH, Johnson LF, Berger FG. Thymidylate synthase overproduction and gene amplification in fluorodeoxyuridine-resistant human cells. Mol Pharmacol. 1985;28:461-7.
- Jenh CH, Geyer PK, Baskin F, Johnson LF. Thymidylate synthase gene amplification in fluorodeoxyuridine-resistant mouse cell lines. Mol Pharmacol. 1985;28:80-5.
- Shimizu T, Nakagawa Y, Takahashi N, Hashimoto S. Thymidylate synthase gene amplification predicts pemetrexed resistance in patients with advanced non-small cell lung cancer. Clin Transl Oncol. 2016;18:107-12.
- Watson RG, Muhale F, Thorne LB, Yu J, O’Neil BH, Hoskins JM, Meyers,MO, Deal AM, Ibrahim JG, Hudson ML, Walko CM, Mcleod HL. & Auman, J. T. Amplification of thymidylate synthetase in metastatic colorectal cancer patients pretreated with 5-fluorouracil-based chemotherapy. Eur J Cancer.2010;46:3358-64.
- Wang TL, Diaz LA Jr, Romans K, Bardelli A. Saha S. Galizia G. Choti M. Donehower R, Parmigiani G, Shih Ie M, Iacobuzio-Donahue C, Kinzler KW, Vogelstein B, Lengauer C, Velculescu, VE. Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients. Proc Natl Acad Sci USA. 2004;101;3089-94.
- Chu J and Dolnick BJ. Natural antisense (rTSalpha) RNA induces site-specific cleavage of thymidylate synthase mRNA. Biochim Biophys Acta. 2002;1587:183-93.
- De Angelis, PM, Svendsrud DH, Kravik KL, Stokke T. Cellular response to 5-fluorouracil (5-FU) in 5-FU-resistant colon cancer cell lines during treatment and recovery. Mol Cancer. 2006;5:20.
- Wang W, Cassidy J, O’brien V, Ryan KM, Collie-Duguid E. Mechanistic and predictive profiling of 5-Fluorouracil resistance in human cancer cells. Cancer Res. 2004b;64:8167-76.
- De Angelis, PM, Fjell B, Kravik KL, Haug T, Tunheim SH, Reichelt W, Beigi M, Clausen OP, Galteland E, Stokke T. Molecular characterizations of derivatives of HCT116 colorectal cancer cells that are resistant to the chemotherapeutic agent 5-fluorouracil. Int J Oncol. 2004;24:1279-88.
- Kang HC, Kim IJ, Park HW, Jang SG, Ahn SA, Yoon SN, Chang HJ, Yoo BC, Park JG. Regulation of MDK expression in human cancer cells modulates sensitivities to various anticancer drugs: MDK overexpression confers to a multi-drug resistance. Cancer Lett. 2007;247:40-7.
- Schmidt WM, Kalipciyan M, Dornstauder E, Rizovski B, Steger GG, Sedivy R, Mueller MW, Mader, RM. Dissecting progressive stages of 5-fluorouracil resistance in vitro using RNA expression profiling. Int J Cancer. 2004;112;200-12.
- Schmidt WM, Kalipciyan M, Dornstauder E, Rizovski B, Sedivy R, Steger GG, Muller MW, Mader RM. Gene expression profiling of colon cancer reveals a broad molecular repertoire in 5-fluorouracil resistance. Int J Clin Pharmacol Ther. 2003;41:624-5.
- Szoke D, Gyorffy A, Surowiak P, Tulassay Z, Dietel M, Gyorffy B. Identification of consensus genes and key regulatory elements in 5-fluorouracil resistance in gastric and colon cancer. Onkologie. 2007;30:421-6.