Developing antibody therapeutics
Posted: 10 June 2015 | David Matthews (MRC Technology)
Monoclonal antibodies have come of age as therapeutics; there are now more than 30 antibodies on the market and over 200 in clinical trials. The attractions of therapeutic antibodies lie not only in their potential to make potent drugs but also their relative low risk in comparison to small molecule programs.
Current data suggest that around 80% of antibody therapeutic pass Phase I clinical trials, 50% pass phase II and 75% pass Phase III2 and this compares favourably with small molecule drug discovery. They also make money: Humira (adalimumab, Abbvie), a treatment for rheumatoid arthritis, is predicted to generate nearly $10 billion by 20163. This article provides a snap shot of the antibody therapeutics field. For a more in-depth review and explanation of the field, I recommend the book Therapeutic Antibody Engineering by Bill Strohl3.
Humanisation model
There are a number of ways of generating therapeutic monoclonal antibodies. Humanisation using monoclonal antibodies from rodents and more recently from the rabbit was the first technical breakthrough and humanisation remains an important technique to reduce the immunogenicity of non-human antibodies. This clinically-validated technology has been applied to a number of successful therapeutics including trastuzumab (Herceptin®)4, natalizumab (Tysabri®)5 and more recently, pembrolizumab (Keytruda®)6. Humanisation is the conversion of a monoclonal antibody derived from a non-human source, typically from a mouse, into a human antibody, which is shown schematically in Figure 1 (page 28). The structure of mammalian antibodies is well conserved between species and fortuitously for us this is especially true between rodents and humans.
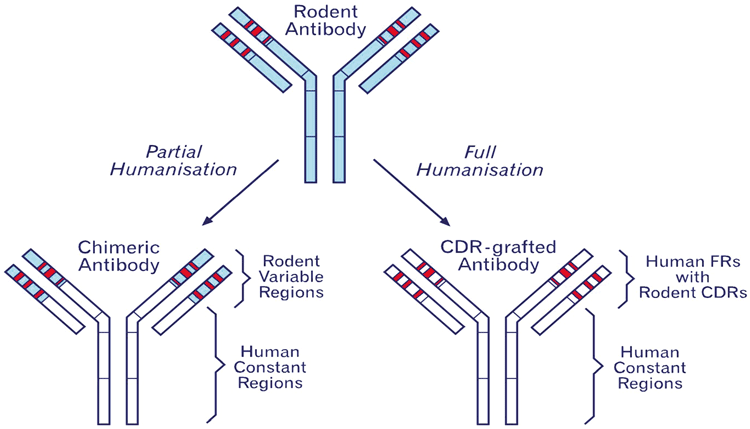
Figure 1: A schematic diagram of humanisation. Complementary determining regions (CDRs) are shown in red and are surrounded by framework (FR). The blue areas indicate rodent sequences. The process of humanisation attempts to limit the murine sequence to the CDRs and essential framework residues in order to minimise immunogenicity.
The antibody’s structure is split into variable domains and the constant domains as shown in Figure 1 and Figure 2. The variable domains each contain three hypervariable complementary determining regions (CDRs) that in three dimensional space form a binding region. The CDRs’ structure is supported by the surrounding framework regions and humanisation is in essence the process of identifying human framework regions that closely match the mouse sequence and swapping in the rodent CDRs. The technique of CDR grafting was first described by Greg Winter’s Group in 19887 and has been refined to a reliable process where, at least in our hands, we would expect to retain 90-100% of binding8.
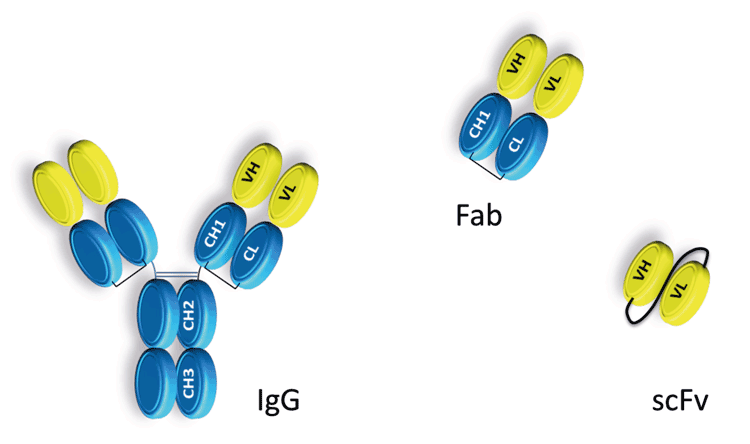
Figure 2: A schematic representation of whole IgG antibody and antibody fragments commonly used in drug discovery. IgG antibodies are constructed from a variable domain shown in yellow and a constant domain depicted in blue. Fc effector ADCC and CDC function is dependent on the hinge region located between the CH1 and CH2 domains and the glycosylation of residue Asn 297.
Phage display
As companies have attempted to improve and industrialise antibody therapeutics, other technologies have come to dominate the antibody field, such as antibody phage display technology. Antibody phage display lends itself to industrialisation and offers the opportunity to more closely monitor and control antibody generation. In most cases commercial phage libraries have been developed from human naive donors 9-12 or even synthetically13,14, obviating the need for humanisation. Whole antibodies cannot be expressed in phage and typically scFv or Fab antibody fragments that contain the V region are displayed on the surface of the bacteriophage. Another advantage of phage display libraries over animal immunisation is that it avoids immunological tolerance, the process by which self-reactive antibodies are removed from the immune response. This is especially important for targets that are highly conserved between species and makes it more likely that antibodies with unique or exquisite properties will be identified and developed.
On the down side, screening hits from naive phage libraries are less likely to have sufficient affinity for drug-like potential and require affinity maturation. However, combined with affinity maturation, phage display is an extremely powerful technique and can drive affinity at extremely high levels in the region of low pM and occasionally to high fM levels.
Transgenics
A third important antibody technology comes in the form of transgenic animals that contain the whole or partial human immunolglobulin loci15,16. Companies such as Mederex and Abgenix pioneered the use of transgenic animals and this has been followed up by Regeneron and others who have added more sophistication to transgenic animal technology by replacing the human constant region with the full repertoire of mouse constant regions17. The main advantage of making a mouse chimeric antibody repertoire is to optimise the mouse B cell response and make it more likely to generate an effective immune response to the immunising antigen. Similar to phage display, transgenic animals offer ‘fully human’ antibodies.
However, there is no clinical evidence that there is any benefit of one technology over the other. Another important consideration is in vivo animal studies. Fully human antibodies may potentially require some form of engineering to reduce immunogenicity for animal in vivo studies longer than one or two weeks or may require the generation of an antibody for mouse studies. The generation of mouse monoclonal antibodies as the starting point does have the advantage of providing the means to conduct in vivo target validation from the outset.
Therapeutic antibody formats
There is a growing variety of therapeutic antibody formats beyond the simple IgG. Of the naturally occurring isotypes ranging from IgM, IgG1,2,3 and 4, IgA and IgE, only IgGs are used for therapeutics and of these IgG1 and IgG4 predominate. These two isotypes offer two distinct functionalities: IgG1 offers potent effector function in the form of antibody dependent cell-mediated cytotoxicity (ADCC) while IgG4 offers the opposite with only residual effector function3. Importantly, IgG1 offers the potential to target a protein, potentially block its activity and recruit an immune response in the form of CD8-positive lymphocytes to target the destruction of the target cell. Examples of drugs using this approach are alemtuzumab (Campath®/Lemtrada®) 18-20 and rituxumab (Rituxan®/MabThera®/Zytux®) 21 which bind CD52 or CD20, respectively, and induce an ADCC response. The effectiveness of alemtuzumab in depleting CD52 positive cells in the clinic exemplifies the power of the antibody Fc effector function in developing effective biologics.
In contrast, other therapeutics require simply the disruption of an interaction to be effective. Natalizumab (Tysabri®) is an IgG4 humanised monoclonal antibody against alpha-4 (α4) integrin and works by preventing activated T cells from crossing blood vessel walls to reach affected organs22,23. Unlike the case with IgG1, IgG4 binds weakly to Fc gamma receptors since it is less able to recruit a cytotoxic immune response. Therefore, nature has provided two useful frameworks to build biotherapeutics, albeit the fact that they do have limitations.
Engineering the Fc
Although natural antibody isotypes such as IgG1 and IgG4 are effective, this has not prevented antibody engineers attempting to further improve their properties, by increasing half-life or improving stability or effector function. In particular, IgG4 antibodies can be unstable and prone to Fab arm exchange24-26 and this instability can have a serious negative impact in manufacturing. However, IgG4 antibodies can be stabilised by a mutation to the hinge region of the Fc domain S241P 27. Further enhancements can be made by improving the binding to the neonatal Fc receptor, FcRn, which has been shown to improve half-life in vivo 28. This has been achieved by introducing mutations into the Fc region that enhance the affinity between the Fc and FcRn. This can extend the half-life by up to three-fold of an IgG1 molecule from the typical 21 days29.
Antibody-dependent cellular cytotoxicity
The third area engineers have focused on is improving effector function either with antibody-dependent cellular cytotoxicity (ADCC) or indeed with complement-dependent cytotoxicity (CDC). Similar to half-life extension, mutations can be introduced to heavy chain constant regions or alternatively through glyco-modification of Fc-linked oligosaccharides3. The major interaction sites of the Fc to the effector Fc gamma receptor ligands are in the hinge and CH2 regions and the binding is dependent on the glycoform of the oligosaccharides attached at residue Asn 297 in the CH2 domain. Improvements in Fc receptor binding can have profound improvements in ADCC activity ranging from 10-100 fold30. Such modifications and enhancements may have significant impact on patients as some individuals have low affinity forms of FcgRIIa and FcgRIIIa and this may result in poorer prognosis in patients treated with biologics such as rituximab.
Despite the effectiveness of antibodies such as rituximab, similar ADCC approaches to solid tumours have been less successful. Since the 1990s there have been attempts to conjugate antibodies with cytotoxic payloads in order to boost the potency of the drug and more recently, the antibody drug conjugate (ADC) field has been reinvigorated; both Genentech (with trastuzumab emtansine31) and Seattle Genetics (with brentuximab vedotin32-35), have successfully brought ADCs to the market and roughly 30 more in ADCs currently in clinical trials. However ADC offer substantial development challenges in comparison to so called ‘naked’ antibodies. The success of ADC depends on a number of factors dependent on both the target and the antibody. To summarise, ADCs require the target cell to internalise on antibody binding in order to deliver the cytotoxic agent into the cell. The target protein must be expressed at a high enough level and internalise at a sufficient rate in order to deliver a therapeutic dose. In addition the antibody must be able to withstand the conjugation chemistry and remain active while attached to a large hydrophobic moiety. As of yet there does not appear to be any guidelines to determine which antibodies are capable of withstanding conjugation other than determining this by trial and error.
A successful example of an ADC is trastuzumab emtansine developed by Genentech with Immunogen31. This is the humanised monoclonal antibody trastuzumab conjugated with the maytansinoid, DM1, a potent microtubule-disrupting agent, joined by a stable linker. The target protein is Her2, the conjugate is internalised, and an active derivative of DM1 is subsequently released by proteolytic degradation of the antibody within the lysosome. Although trastuzumab has had a major impact on the treatment of Her2-positive breast cancers, Her2-positive patients that do not respond to treatment remain, and ultimately most tumours develop resistance to the antibody despite continuing to express Her236. The EMILIA clinical phase III trial of patients already resistant to trastuzumab data suggests that trastuzumab emtansine is effective and prolonged life by 5.8 months compared to the combination of lapatinib and capecitabine37. Therefore, trastuzumab emtansine and brentuximab vedotin have both validated the ADC approach and has showed the value of the ADC approach.
Despite the progress of ADC, significant challenges remain and include improvement of the therapeutic index, linked to a careful selection of the targets, a better understanding of ADC mechanism of action and the management and understanding of ADC off-target toxicities.
Bispecifics
An alternative to the ADC approach is developing bi–specific antibodies, where two antibody-like molecules are brought together, fused or chemically linked, to add extra functionality. An example of this is Micromet AG’s BiTE™ technology developed in order to enhance the cytotoxic potency of biotherapeutics. Blinatumomab38-41 is perhaps the most advanced example where the BiTE is constructed from two single-chain variable fragments (scFvs) of different antibodies for CD3 and CD19. The anti-CD3 scFv binds to T cells via the CD3 receptor, and the other to a tumour cell – in this case CD19-positive B cell acute lymphoblastic leukaemia. The result is cytotoxic T cell activation directed to the target B cells. Amgen, who subsequently purchased Micromet, have taken blinatumomab to market and received approval in the US in December 2014. However, the BiTE™ technology is only one of many differing bi-specific formats42, that include but are not limited to: heteromeric IgGs, Fabs and scFv, nanobodies such as VHH and combinations thereof.
One interesting format is the dual variable domain (DVD) antibody where the V regions of one antibody is transplanted on top of another as a series of V regions43. The fact that this is possible and binding for both antibodies retained is a testament to the stability of the basic antibody structure. The advantages of bi-specifics are they require lower dosing. In addition, unlike with ADCs, targets are not required to be highly expressed but there still remain challenges. Manufacturing and poor expression has held back some formats and for some target combinations assessing the desired affinity of each antibody molecule can result in complex pharmacology that can only be explored in vivo.
Implications for antibody drug development
The question one might now ask is: do plain old normal antibodies have a future as therapeutics and in order to develop successful drugs do we need access to the wide variety of technologies now available? Pharma has access to most of the antibody technology it requires, but for smaller organisations and biotechs the issue is how to plot a cost effective route through antibody drug development. The attraction of technologies such as ADCs and bi-specific approaches such as the BiTE™ format is the enhanced cytotoxicity and are platform technologies like these essential and is there any place for plain old vanilla antibodies. The answer I think is yes and a good example of this is the development of Pembroluzumab, an antibody that blocks the interaction between PD-1 and its ligands. The murine antibody was initially developed by Organon Pharma and the antibody was generated using old fashioned hybridoma technology. Subsequently Organon used MRC Technology humanise the antibody and Organon then went on to partner the antibody with Schering Plough Research Institute and then onto Merck & Co who took the antibody through to market approval in late 2014 for melanoma. What is important about this story is that Pembroluzumab success it relied on good science and project management not a platform technology. The conclusion is that there is no technological barrier for research groups and small biotech to develop credible antibody therapeutics.
References
- Reichert, J. M. Antibodies to watch in 2015. mAbs 7, 1-8, doi:10.4161/19420862.2015.988944 (2015).
- Reichert, J. M. Metrics for antibody therapeutics development. mAbs 2, 695-700 (2010)
- Strohl W., R. & Strohl L., M. Therapeutic Antibody Engineering. (Woodhead Publishing, 2012)
- Hudis, C. A. Trastuzumab–mechanism of action and use in clinical practice. The New England journal of medicine 357, 39-51, doi:10.1056/NEJMra043186 (2007)
- Polman, C. H. et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. The New England journal of medicine 354, 899-910, doi:10.1056/NEJMoa044397 (2006)
- Poole, R. M. Pembrolizumab: first global approval. Drugs 74, 1973-1981, doi:10.1007/s40265-014-0314-5 (2014)
- Riechmann, L., Clark, M., Waldmann, H. & Winter, G. Reshaping human antibodies for therapy. Nature 332, 323-327, doi:10.1038/332323a0 (1988)
- Williams D.G., Matthews D.J. & T, J. Humanising Antibodies by CDR Grafting. Vol. 1 319-339 (Springer-Verlag Berlin Heidelberg, 2010)
- Griffiths, A. D. et al. Isolation of high affinity human antibodies directly from large synthetic repertoires. The EMBO journal 13, 3245-3260 (1994)
- Barbas, C. F., 3rd et al. Recombinant human Fab fragments neutralize human type 1 immunodeficiency virus in vitro. Proceedings of the National Academy of Sciences of the United States of America 89, 9339-9343 (1992)
- Marks, J. D. et al. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. Journal of molecular biology 222, 581-597 (1991)
- Vaughan, T. J. et al. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nature biotechnology 14, 309-314, doi:10.1038/nbt0396-309 (1996)
- Sidhu, S. S. et al. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. Journal of molecular biology 338, 299-310, doi:10.1016/j.jmb.2004.02.050 (2004)
- Nelson, B. & Sidhu, S. S. Synthetic antibody libraries. Methods in molecular biology 899, 27-41, doi:10.1007/978-1-61779-921-1_2 (2012)
- Green, L. L. Antibody engineering via genetic engineering of the mouse: XenoMouse strains are a vehicle for the facile generation of therapeutic human monoclonal antibodies. Journal of immunological methods 231, 11-23 (1999)
- Lonberg, N. Human antibodies from transgenic animals. Nature biotechnology 23, 1117-1125, doi:10.1038/nbt1135 (2005)
- Dechiara, T. M. et al. VelociMouse: fully ES cell-derived F0-generation mice obtained from the injection of ES cells into eight-cell-stage embryos. Methods in molecular biology 530, 311-324, doi:10.1007/978-1-59745-471-1_16 (2009)
- Crowe, J. S., Hall, V. S., Smith, M. A., Cooper, H. J. & Tite, J. P. Humanized monoclonal antibody CAMPATH-1H: myeloma cell expression of genomic constructs, nucleotide sequence of cDNA constructs and comparison of effector mechanisms of myeloma and Chinese hamster ovary cell-derived material. Clinical and experimental immunology 87, 105-110 (1992)
- Dyer, M. J., Hale, G., Hayhoe, F. G. & Waldmann, H. Effects of CAMPATH-1 antibodies in vivo in patients with lymphoid malignancies: influence of antibody isotype. Blood 73, 1431-1439 (1989)
- Gilleece, M. H. & Dexter, T. M. Effect of Campath-1H antibody on human hematopoietic progenitors in vitro. Blood 82, 807-812 (1993)
- de Haij, S. et al. In vivo cytotoxicity of type I CD20 antibodies critically depends on Fc receptor ITAM signaling. Cancer research 70, 3209-3217, doi:10.1158/0008-5472.CAN-09-4109 (2010)
- O’Connor, P. et al. Relapse rates and enhancing lesions in a phase II trial of natalizumab in multiple sclerosis. Multiple sclerosis 11, 568-572 (2005)
- Rice, G. P., Hartung, H. P. & Calabresi, P. A. Anti-alpha4 integrin therapy for multiple sclerosis: mechanisms and rationale. Neurology 64, 1336-1342, doi:10.1212/01.WNL.0000158329.30470.D0 (2005)
- Aalberse, R. C. & Schuurman, J. IgG4 breaking the rules. Immunology 105, 9-19 (2002)
- Labrijn, A. F. et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nature biotechnology 27, 767-771, doi:10.1038/nbt.1553 (2009)
- Rispens, T. et al. Human IgG4 binds to IgG4 and conformationally altered IgG1 via Fc-Fc interactions. Journal of immunology 182, 4275-4281, doi:10.4049/jimmunol.0804338 (2009)
- Angal, S. et al. A single amino acid substitution abolishes the heterogeneity of chimeric mouse/human (IgG4) antibody. Molecular immunology 30, 105-108 (1993)
- Datta-Mannan, A. et al. Humanized IgG1 variants with differential binding properties to the neonatal Fc receptor: relationship to pharmacokinetics in mice and primates. Drug metabolism and disposition: the biological fate of chemicals 35, 86-94, doi:10.1124/dmd.106.011734 (2007)
- Zalevsky, J. et al. Enhanced antibody half-life improves in vivo activity. Nature biotechnology 28, 157-159, doi:10.1038/nbt.1601 (2010)
- Presta, L. G. Molecular engineering and design of therapeutic antibodies. Current opinion in immunology 20, 460-470, doi:10.1016/j.coi.2008.06.012 (2008)
- Lambert, J. M. & Chari, R. V. Ado-trastuzumab Emtansine (T-DM1): an antibody-drug conjugate (ADC) for HER2-positive breast cancer. Journal of medicinal chemistry 57, 6949-6964, doi:10.1021/jm500766w (2014)
- Chen, R., Wang, F., Zhang, H. & Chen, B. Brentuximab vedotin for treatment of relapsed or refractory malignant lymphoma: results of a systematic review and meta-analysis of prospective studies. Drug design, development and therapy 9, 2277-2283, doi:10.2147/DDDT.S83592 (2015)
- Graf, S. A. & Gopal, A. K. Treatment of relapsed classical Hodgkin lymphoma in the brentuximab vedotin era. Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Education Program 2014, 151-157, doi:10.1182/asheducation-2014.1.151 (2014)
- Koh, K. N. et al. Successful use of brentuximab vedotin for refractory anaplastic large cell lymphoma as a bridging therapy to haploidentical stem cell transplantation and maintenance therapy post-transplantation. Pediatric blood & cancer 62, 1063-1065, doi:10.1002/pbc.25351 (2015)
- Moskowitz, C. H. et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet, doi:10.1016/S0140-6736(15)60165-9 (2015)
- Nahta, R. & Esteva, F. J. HER-2-targeted therapy: lessons learned and future directions. Clinical cancer research : an official journal of the American Association for Cancer Research 9, 5078-5084 (2003)
- Krop, I. E. et al. Trastuzumab emtansine (T-DM1) versus lapatinib plus capecitabine in patients with HER2-positive metastatic breast cancer and central nervous system metastases: a retrospective, exploratory analysis in EMILIA. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO 26, 113-119, doi:10.1093/annonc/mdu486 (2015)
- Advani, A. S. Blinatumomab: a novel agent to treat minimal residual disease in patients with acute lymphoblastic leukemia. Clinical advances in hematology & oncology : H&O 9, 776-777 (2011)
- Bassan, R. Toward victory in adult ALL: blinatumomab joins in. Blood 120, 5094-5095, doi:10.1182/blood-2012-10-460394 (2012)
- Thomas, X. Blinatumomab: a new era of treatment for adult ALL? The Lancet. Oncology 16, 6-7, doi:10.1016/S1470-2045(14)71183-0 (2015)
- Zimmerman, Z., Maniar, T. & Nagorsen, D. Unleashing the clinical power of T cells: CD19/CD3 bi-specific T cell engager (BiTE(R)) antibody construct blinatumomab as a potential therapy. International immunology 27, 31-37, doi:10.1093/intimm/dxu089 (2015)
- Kontermann, R. E. & Brinkmann, U. Bispecific antibodies. Drug discovery today, doi:10.1016/j.drudis.2015.02.008 (2015)
- Gu, J. & Ghayur, T. Generation of dual-variable-domain immunoglobulin molecules for dual-specific targeting. Methods in enzymology 502, 25-41, doi:10.1016/B978-0-12-416039-2.00002-1 (2012)
Biography
David Matthews did his PhD in immunology at University College London studying the functional relationship of IL-4 and IL-13. He subsequently did post docs at Wurzburg University and at the LMB in Cambridge, UK, again studying the functional roles of Il-4 and IL-13. In 2001 he joined AERES Biomedical Ltd, a biotech specialising in antibody humanisation. In 2006 he joined the MRC Technology antibody engineering group and in 2012 he became the head of the BioTherapeutics Group. David and his team have been developing therapeutic antibodies to a wide range of targets in collaboration with academic and industrial partners.
Related topics
Antibody Discovery, Drug Discovery, Immunogenicity
Related organisations
MRC Technology
Related people
David Matthews