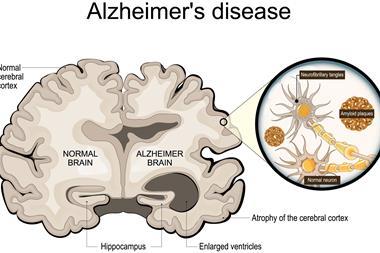
Whole genome sequencing identified 17 significant variants associated with AD risk in five genomic regions.

Researchers at the Boston University School of Public Health (BUSPH) and UTHealth Houston School of Public Health have found multiple genetic variants that may influence Alzheimer’s disease (AD) risk, progressing the understanding of biological pathways to target.
Whole genome sequencing was used, which identified 17 significant variants associated with AD in five genomic regions. This data builds upon genome-wide association studies, which only concentrate on common variants and regions, allowing the team to pinpoint rare and important genes and variants.
Dr Anita DeStefano, professor of biostatistics at BUSPH, explained: “Prior genome-wide association studies using common variants have identified regions of the genome, and sometimes genes, that are associated with Alzheimer’s disease…Whole genome sequence data interrogates every base pair in the human genome and can provide more information about which specific genetic change in a region may be contributing to Alzheimer’s disease risk or protection.”
In the study, the researchers conducted single variant association analyses and rare variant aggregation association tests using whole genome sequencing data from the Alzheimer’s Disease Sequencing Project (ADSP). The ADSP data includes over 95 million variants among 4,567 participants with or without the disease. The KAT8 variant was one of the most notable among the 17 significant variants linked to AD. It was associated with the disease in both the single and rare variant analyses, including several rare TREM2 variants.
Co-lead and corresponding author Dr Chloé Sarnowski, assistant professor in the Department of Epidemiology at UTHealth Houston School of Public Health, commented: “By using whole genome sequencing in a diverse sample, we were able to not only identify novel genetic variants associated with Alzheimer’s disease risk in known genetic regions, but also characterise whether the known and novel associations are shared across populations.”
Historically, Black and Latino populations have been underrepresented in genetic studies of AD despite having a higher prevalence of the disease than other ethnic groups. Therefore, the ADSP includes ethnically diverse participants, and the population-specific assessments focused on White/European-ancestry, Black/African-American, and Hispanic/Latino subgroups, as well as a multi-population meta-analysis.
“Including participants that represent diverse genetic ancestry and diverse environments in terms of social determinants of health is important to understanding the full spectrum of Alzheimer’s disease risk, as both the prevalence of the disease and the frequencies of genetic variants can differ among populations,” Dr DeStefano added.
The team had limited ability to detect associations because the sample sizes in the population-specific analyses were small. However, Dr DeStefano said: “we replicated known population differences for the APOE gene, which is one of the best-known and strongest risk genes for Alzheimer’s disease.”
Moving forward, the researchers aim to expand the research to use whole genome sequencing with larger sample sizes in the ADSP and investigate how variants affect biological functioning.
This study was published in Alzheimer’s & Dementia: The Journal of the Alzheimer's Association.