

New generative AI method could make drug discovery faster
Posted: 23 January 2026 | Drug Target Review | No comments yet
A newly developed AI-driven technique could dramatically speed up the discovery of drugs and advanced materials, enabling scientists to design chemically valid, property-targeted molecules in minutes rather than years.

A new artificial intelligence technique could significantly accelerate the discovery of new drugs and materials, cutting years from a process that traditionally involves time consuming trial and error.
The research from the University of Florida and New York University outlines a method that can generate promising molecular candidates around 10 times faster than existing AI approaches, without sacrificing accuracy or chemical realism. The work highlights how generative AI is beginning to reshape the foundations of chemistry and materials science.
From educated guesses to guided design
Every modern medicine and advanced material begins as a hypothesis: a proposed arrangement of atoms that might kill bacteria, store energy or interact efficiently with light. With billions of possible small molecules to choose from, identifying the right one has long been a near impossible problem.
Rather than modifying known compounds, AI models can invent entirely new molecular structures based on desired properties.
Recent advances in generative AI have helped narrow that search. Rather than modifying known compounds, AI models can invent entirely new molecular structures based on desired properties. The newly developed method, called PropMolFlow (Property-guided Molecular Flow), pushes this approach further by dramatically increasing the speed at which viable candidates can be generated.
“For most of scientific history, material discovery often preceded understanding – useful compounds were found by accident, then scientists figured out why they worked,” says Stefano Martiniani, Assistant Professor of Physics, Chemistry, Mathematics and Neural Science at NYU and an author of the paper. “Generative AI offers the possibility of inverting this: specify the properties, then find the structures. PropMolFlow represents another step toward making that vision practical.”
Designing molecules backwards
The researchers describe molecular design as an ‘inverse problem’. Scientists are rarely interested in a molecule for its own sake; instead, they want one that performs a specific function.
“Chemists don’t usually want ‘a molecule,’” explains Martiniani. “Instead, they want a molecule that does something specific – to interact strongly with light for optical applications or to possess a particular electronic structure that determines how it absorbs energy or conducts electricity.”
PropMolFlow builds on earlier AI models inspired by image-generation tools such as DALL-E, which were first adapted to molecular design in 2022.
PropMolFlow builds on earlier AI models inspired by image-generation tools such as DALL-E, which were first adapted to molecular design in 2022. While previous approaches improved accuracy or chemical validity, they often required thousands of computational steps. PropMolFlow achieves similar or better results in roughly 100 steps.
“For a field where computational speed directly translates to discovery speed, this represents a meaningful advance,” adds Mingjie Liu, Assistant Professor in the University of Florida’s Department of Chemistry and one of the paper’s authors. “The work doesn’t replace what came before it, but, rather, demonstrates that the next generation of molecular generators can be substantially faster while maintaining the accuracy that makes these tools useful.”
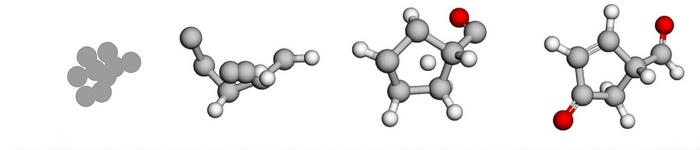
A new AI model designs molecules with specified properties 10 times faster than previous methods, potentially speeding up the process for the creation of pharmaceuticals and materials. The figure illustrates how the system transforms random noise into complete molecular structures guided by target properties. (Image courtesy of the University of Florida and New York University.)
Accuracy without shortcuts
Speed alone is not enough if generated molecules break basic chemical rules. The team therefore tested PropMolFlow against established models, finding that it produced chemically valid structures more than 90 percent of the time.
Speed alone is not enough if generated molecules break basic chemical rules.
“This matters because many earlier approaches produced structures that looked superficially plausible but violated basic chemical rules,” says Martiniani.
To avoid the risk of “AI grading its own homework”, the researchers validated their results using density functional theory, a physics-based method that does not rely on machine learning.
“This kind of validation provides the credibility needed for generated molecules to be taken seriously for real applications,” says Liu.
Implications for discovery
The researchers believe the combination of speed, accuracy and rigorous validation could be transformational for early-stage molecular discovery.
“With the ability to generate thousands of chemically valid, property-targeted candidates in minutes rather than hours, researchers can iterate faster,” Martiniani explains.
While extending the approach to larger, more complex molecules remains a challenge, Liu notes that the principles behind PropMolFlow provide “a template for more ambitious applications” in drug development and advanced materials research.
Related topics
Artificial Intelligence, Computational techniques, Drug Discovery, Drug Discovery Processes, Molecular Modelling, Small Molecules
Related organisations
Florida University, New York University