Huntington's disease remains a major challenge, but allele-selective gene editing offers new hope. By targeting only the mutant gene, this approach could provide a one-time, durable treatment. Life Edit’s Dr Amy Pooler reveals how this innovative therapy could reshape HD treatment.
Huntington's disease (HD) is a devastating neurodegenerative disorder with profound generational impacts. As an autosomal dominant condition that typically manifests in adulthood, HD affects entire families across multiple generations, with each child of an affected parent facing a 50 percent chance of inheriting the disease-causing mutation. This hereditary pattern makes HD particularly challenging from both medical and societal perspectives: a condition with a known genetic cause but, to date, no disease-modifying treatments.
At Life Edit, the gene editing and R&D technology business of ElevateBio, we are advancing gene editing technologies that offer new hope for addressing this condition at its source. Our development of an allele-selective gene editing approach represents the potential to fundamentally alter the HD treatment landscape through a one-time intervention designed to provide durable benefit, rather than requiring chronic administration. This combination of single-dose treatment with allele selectivity represents a unique approach among investigational HD therapies in development.
Understanding Huntington's disease
Huntington's disease is a rare, inherited neurodegenerative disorder affecting approximately 41,000 people in the US, with another 200,000 at risk of developing the condition.1,2 The disease manifests in mid-adulthood with a progressive triad of motor dysfunction, behavioural abnormalities and cognitive decline. What makes HD particularly devastating is its predictable trajectory – symptoms typically appear between ages 40-50 and progressively worsen.3
Huntington’s disease is a rare, inherited neurodegenerative disorder affecting approximately 41,000 people in the US, with another 200,000 at risk of developing the condition
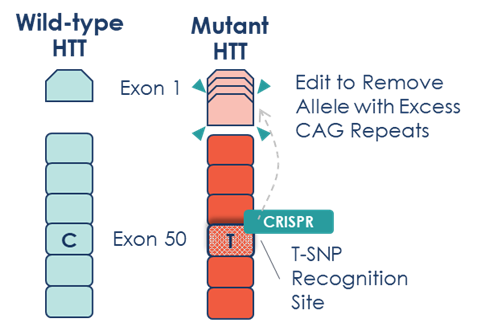
At its core, HD is caused by a CAG (cytosine, adenine, guanine) trinucleotide repeat expansion in the huntingtin gene (HTT). While everyone carries two copies of the HTT gene, individuals with HD have inherited a mutated copy containing an abnormally long stretch of these CAG repeats. This genetic anomaly results in the production of a mutant huntingtin protein (mHTT) that aggregates in neurons, causing cellular dysfunction and eventually neuronal death. The progression of damage is particularly pronounced in the striatum and cortex, regions critical for motor control and cognitive function.
The dilemma of huntingtin suppression
Previous therapeutic approaches for HD have focused on lowering overall huntingtin levels. However, this strategy presents a fundamental challenge. While reducing mutant huntingtin is desirable, the wild-type huntingtin protein plays crucial roles in normal neuronal function, including vesicular transport, synaptic function and transcriptional regulation. Thus complete suppression of all huntingtin protein could potentially create new problems despite addressing the original disease.
This dilemma has driven our research at Life Edit towards allele-selective approaches that target only the toxic mutant huntingtin gene while preserving the healthy copy – a precision medicine approach that addresses the disease mechanism while minimising potential adverse effects.
Life Edit's novel approach: LETI-101
Our research team at Life Edit has made significant progress in developing allele-selective editing approaches for HD. Our lead Huntington's disease programme, LETI-101, represents a novel approach to allele-selective editing using advanced CRISPR technology.
What makes our LETI-101 approach distinctive is its targeting strategy. Rather than targeting the CAG repeat expansion directly, we leverage a single nucleotide polymorphism (SNP) – small genetic variations that differ between the mutant and healthy alleles in many HD patients.
Specifically, LETI-101 targets the 'T' allele of an exonic SNP (rs362331) in the HTT gene. For patients who are heterozygous for this SNP (carrying one 'C' and one 'T' allele), this approach allows for selective editing of only the mutant copy of HTT.

Figure 1: Life Edit’s allele-selective approach recognises the difference between wild-type HTT and mutant HTT by targeting a specific T-SNP recognition site in Exon 50, allowing selective editing of only the disease-causing gene.[/caption]
A transformative aspect of our LETI-101 approach is that it's designed as a one-time treatment that could provide long-lasting benefit without the need for repeated administration. Unlike other therapeutic modalities being developed for HD that would require ongoing treatment to maintain efficacy, our gene editing therapy is intended to make a permanent, precise modification to the DNA itself.
This fundamentally different approach – combining the durability of a single-dose therapy with the precision of allele-selective editing – addresses a critical unmet need in HD treatment. By potentially eliminating the burden of chronic therapy while selectively targeting only the disease-causing gene variant, LETI-101 offers distinct potential advantages in both efficacy and convenience for patients.
The technology behind LETI-101
At the heart of LETI-101's functionality is our specialised gene editing technology that overcomes key limitations of conventional CRISPR systems. We have developed compact nucleases that are smaller than standard CRISPR tools, enabling the complete editing system to fit within a single viral delivery vector.

Figure 2: LETI-101 is a complete gene editing system with Life Edit’s proprietary, compact LEG-B nuclease, packaged in an AAV5 vector for all-in-one delivery to the brain.[/caption]
The ability to recognise diverse DNA sequences required for binding, called protospacer adjacent motif or ‘PAM’ sequences, makes our approach particularly powerful. This versatility allows our technology to access genetic locations that would otherwise be inaccessible to other CRISPR systems. In LETI-101, our engineered nuclease specifically recognises the sequence created by the presence of the 'T' allele at the rs362331 SNP site, enabling the highly selective targeting that distinguishes healthy from mutant huntingtin genes. The editing systems create allele-specific double-strand breaks – distinct insertion and deletion mutations knowns as ‘indels’ – that activate cellular repair pathways, creating frameshift mutations that lead to degradation of the mutant mRNA.
Furthermore, we chose delivery via adeno-associated viral vector (AAV) given the extensive evidence supporting its use in the central nervous system (CNS). It has been shown to result in significant levels of brain cell transduction, which is important for maximising potential therapeutic benefit.
From lab to potential therapy: the evidence for LETI-101
To identify editing systems targeting the mutant HTT gene, we performed initial studies in cultured fibroblast cells derived from three different individuals affected by HD who were all heterozygous for the rs362331 SNP. We observed efficient editing of mutant HTT as determined by the presence of CRISPR-induced indels and complete allele specificity, with editing exclusively in the 'T' allele. This resulted in approximately 55 percent selective reduction of mutant huntingtin protein while fully preserving the wild-type protein.
Next, we tested the performance of this editing system in an animal model of Huntington’s disease. We selected BACHD transgenic mice for this study, since they carry a full-length human mutant HTT gene with the targeted exon50 'T' SNP. To deliver our CRISPR editing system to the brain cells involved in the disease, we packaged it into AAV to make LETI-101. When infused into the striatum, a brain region critically affected by HD, LETI-101 showed strong dose-dependent effects. We observed robust AAV vector distribution across key brain regions along with significant guide RNA and nuclease expression. Most importantly, the treatment achieved up to 80 percent reduction of mutant huntingtin protein in the striatum, with substantial reductions also observed in the thalamus and cortex. These reductions significantly exceed the 40 percent threshold considered necessary for clinical relevance, suggesting potential therapeutic benefit.
Our safety studies in healthy adult cynomolgus monkeys were equally promising. No adverse effects were observed, even at the highest dose, and at all doses tested we observed potentially clinically-relevant transgene expression across key HD-relevant brain regions, minimal systemic vector distribution, and no detectable immune response to the nuclease. We utilised MRI-guided convection-enhanced delivery, demonstrating potential clinical feasibility of targeted delivery to the striatum.
Broader platform potential
While LETI-101 represents a significant advancement for Huntington's disease specifically, our technology platform has broader implications. The diversity of nucleases with varied PAM recognition sequences in our collection enables targeted approaches for many other genetic diseases.

Figure 3: Life Edit’s nuclease collection includes a broad range of genomic recognition sites – also known as protospacer adjacent motifs (PAMs) – which determine the DNA segments in the genome to which a nuclease can bind to edit a gene. Life Edit’s diverse library increases the number of sites where therapeutically meaningful edits can be made, expanding the potential application of its gene editing platform.[/caption]
We believe the ability to selectively target disease-causing alleles while preserving healthy genes could potentially revolutionise the treatment paradigm for numerous monogenic disorders, particularly those where complete gene knockouts would be detrimental.
The road ahead
We recently presented data at the 20th Annual Huntington's Disease Therapeutics Conference in February 2025 that provides compelling evidence for LETI-101's potential efficacy and safety across multiple experimental systems.4
Through our research, we have established clear success metrics: potent repression of mutant huntingtin in the striatum with preservation of the essential wild-type huntingtin protein. Additionally, our team has already gained alignment with the UK's Medicines and Healthcare Products Regulatory Agency (MHRA) on its development strategy. Taken together, the compelling preclinical results and favourable feedback from the MHRA on our chemistry, manufacturing & controls (CMC) and development path, strongly support LETI-101’s advancement as a development candidate.
We believe that combining Life Edit's gene editing technology with ElevateBio's manufacturing capabilities creates a unique value proposition for potential partners looking to advance cutting-edge genetic medicines for HD, with LETI-101 now positioned to begin the final studies required before human clinical trials. More importantly, LETI-101 is a promising programme that has the potential to become a transformative, disease-modifying therapy for individuals living with Huntington’s disease.
Conclusion
The progression of LETI-101 from concept to development candidate illustrates the potential of precision gene editing approaches for previously untreatable neurodegenerative disorders. By selectively targeting the mutant huntingtin allele while preserving the essential wild-type protein, we're addressing a fundamental challenge in HD therapeutics.
This advancement validates not only the gene editing technology itself but also demonstrates how an integrated approach connecting advanced technology, manufacturing capabilities and therapeutic development can accelerate progress towards treating devastating genetic diseases. The rapid progression from concept to a development candidate poised for clinical studies exemplifies the potential of this comprehensive approach to change the future of medicine.
References
1. Yohrling G, Raimundo K, Crowell V, Lovecky D, Vetter L, Seeberger L. "Prevalence of Huntington's Disease in the US (954)." Neurology. April 14, 2020; 94(15_supplement). https://doi.org/10.1212/WNL.94.15_supplement.954
2. Huntington's Disease Society of America. "What is Huntington's Disease?" HDSA.org. Accessed March 2025. https://hdsa.org/what-is-hd/
3. Huntington's Disease Society of America. "Huntington's Disease Symptoms" HDSA.org. Accessed March 2025. https://hdsa.org/what-is-hd/huntingtons-disease-symptoms/
4. Brown LY, et al. "AAV5-delivered Life Edit CRISPR system results in broad CNS biodistribution and allele selective editing and reduction of mutant HTT protein in critical HD brain regions." Presented at the 20th Annual Huntington's Disease Therapeutics Conference, February 24-27, 2025, Palm Springs, CA.
About the author
 Dr Amy Pooler is Senior Vice President of Research & Development at Life Edit, ElevateBio’s gene editing and R&D technology business. She has more than 20 years of scientific leadership and expertise with an extensive background in genetic medicine, neuroscience, drug development, and scientific strategy. Life Edit is seeking collaborations to accelerate LETI-101's development.
Dr Amy Pooler is Senior Vice President of Research & Development at Life Edit, ElevateBio’s gene editing and R&D technology business. She has more than 20 years of scientific leadership and expertise with an extensive background in genetic medicine, neuroscience, drug development, and scientific strategy. Life Edit is seeking collaborations to accelerate LETI-101's development.
Topics
- Analytical Techniques
- Cancer
- CRISPR
- Dr Amy Pooler (Senior Vice President of Research & Development at Life Edit)
- Drug Discovery
- Drug Discovery Processes
- Gene Editing Technologies
- Gene Therapy
- Genetic Analysis
- Genome Editing
- Infectious disease
- Legal & Compliance
- Life Edit
- neurodegenerative disorders
- Neurological disorders
- Precision Medicine
- Translational Science