Compound and fragment screening by native mass spectrometry
Posted: 14 June 2019 | Nicola Burgess-Brown, Rod Chalk | No comments yet
Native mass spectrometry has the potential to be a fast, simple, highly sensitive and automatable technique for compound screening in drug design and discovery. In this article, Rod Chalk and Nicola Burgess-Brown discuss the key attributes of native mass spectrometry and reasons why it is not yet widely employed.
Effective and efficient drug discovery is essential if we are to sustain a viable pharmaceuticals industry and meet the medical challenges of the 21st century. Strategies include in silico drug design, discovery from natural products, high-throughput screening of large compound libraries or screening of smaller drug fragment libraries. A variety of screening methods are employed, each with a different propensity to produce unwanted false positive or false negative results.1 Frequently, two or more orthogonal methods are used generating only partially overlapping results, and there is no generally agreed strategy for primary screening and secondary/tertiary validation.
Advantages of native MS
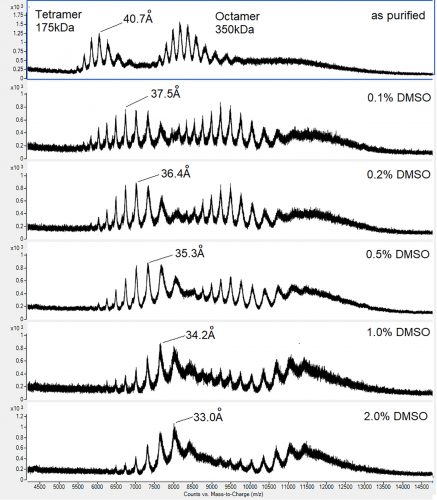
Figure 1: Native m/z spectra for a tetramer-octamer homomultimeric protein complex illustrating the conformational changes induced by different concentrations of DMSO
In principal, native mass spectrometry (MS) is an attractive screening option. Primarily, it is a generic tool that relies on a measurable shift in mass or charge to indicate a binding event, and can thus be applied to any target protein. Contrary to popular opinion, and unlike ion mobility, assignment of charge in electrospray occurs exclusively in the liquid phase,2 and native MS is thus a better reflection of the cellular drug-target interaction than, for example, crystal soaking – a solid phase interaction; or SPR – an interaction at the solid-liquid interface. Since immobilisation is not required, screening artefacts resulting from fusion proteins or chemical conjugates can be avoided. Similarly, artefacts resulting from (expensive) fluorophores are also avoided. Whilst the false negative rate cannot be readily determined, false positives should not theoretically occur. Observation of one mass shift, indicative of a single binding event, is assumed to be specific, whereas multiple binding events indicate lack of specificity. Weak mM interactions are maintained, making the technique suitable for fragment screening, and titration of the compound allows the binding affinity to be determined.3 Protein-protein interactions can be readily measured, as well as the formation or disruption of homomultimeric complexes – an effect that is difficult to detect by other means. Changes in conformation or unfolding resulting from drug binding may also be detected. Robust methods exist for native MS compound screening in the presence of detergents,4 which are essential for drug discovery related to membrane proteins such as GPCRs, representing a significant proportion of all drug targets. Finally, and uniquely, native MS has the capacity for multiplex compound screening. The resolution of the technique is such that mass differences smaller than 1Da can be resolved from a protein-ligand complex. In a thoughtfully designed chemical screen in which isobaric compounds are excluded, multiple interactions can be measured simultaneously, where the identity of each compound is encoded by its mass. Hundreds, and potentially thousands, of compounds can be screened in a single binding experiment, each functioning as an internal control for the others, drastically reducing the time, cost and protein consumption of a screening campaign.
Challenges of native MS
 If the advantages of native MS as a screening tool are so profound, one is forced to question why it is not more widely employed. There are several reasons and understanding them is important in order to make an informed decision as to whether to utilise the technique. Firstly, native MS, like all MS, works only in volatile salt-free buffer. In drug screening, buffer conditions are perceived to be of paramount importance and a typical “physiological” AlphaScreen assay buffer containing 50mM Tris (pH 7.4), 150mM NaCl, 0.05 percent BSA, 0.02 percent Tween, and 1mM DTT is essentially incompatible with the technique. Exchange into a MS-compatible volatile buffer, such as 50mM ammonium acetate pH 6.5, is often assumed to be non-physiological and thus invalid for screening purposes. Our experience, and that of others, indicates this is not generally true. One advantage of the technique is that natively folded protein is readily distinguishable from protein beginning to unfold. Deviations of 0.5pH unit either side of the pH optimum and transfer to salt-free buffer have no discernible effect on the stability or conformation of most proteins. Secondly, instrument vendors have been slow to appreciate the importance of native MS for drug screening. Consequently, although excellent equipment exists, data analysis is comparatively slow and users have been obliged to develop their own software – though recently, commercial third-party software has become available.5 Thirdly, protein consumption in a native MS experiment may be as high as 50μg, making it unsuitable as a primary high-throughput screen where the supply of target protein is limited. Fourthly, while the general level of MS expertise within the pharmaceutical industry is very high, there is far less experience and understanding when applied to native MS. This ranges from misconceptions regarding the equipment required, to disbelief that native MS has any validity at all.
If the advantages of native MS as a screening tool are so profound, one is forced to question why it is not more widely employed. There are several reasons and understanding them is important in order to make an informed decision as to whether to utilise the technique. Firstly, native MS, like all MS, works only in volatile salt-free buffer. In drug screening, buffer conditions are perceived to be of paramount importance and a typical “physiological” AlphaScreen assay buffer containing 50mM Tris (pH 7.4), 150mM NaCl, 0.05 percent BSA, 0.02 percent Tween, and 1mM DTT is essentially incompatible with the technique. Exchange into a MS-compatible volatile buffer, such as 50mM ammonium acetate pH 6.5, is often assumed to be non-physiological and thus invalid for screening purposes. Our experience, and that of others, indicates this is not generally true. One advantage of the technique is that natively folded protein is readily distinguishable from protein beginning to unfold. Deviations of 0.5pH unit either side of the pH optimum and transfer to salt-free buffer have no discernible effect on the stability or conformation of most proteins. Secondly, instrument vendors have been slow to appreciate the importance of native MS for drug screening. Consequently, although excellent equipment exists, data analysis is comparatively slow and users have been obliged to develop their own software – though recently, commercial third-party software has become available.5 Thirdly, protein consumption in a native MS experiment may be as high as 50μg, making it unsuitable as a primary high-throughput screen where the supply of target protein is limited. Fourthly, while the general level of MS expertise within the pharmaceutical industry is very high, there is far less experience and understanding when applied to native MS. This ranges from misconceptions regarding the equipment required, to disbelief that native MS has any validity at all.
Native MS DMSO
When native MS screening has been applied, results do not always confirm those detected using orthogonal methods.6 Native MS is not unique in this respect, but one aspect of the method, the use of DMSO, may be responsible. Since many compounds are both soluble and stable in DMSO, and it is relatively non-toxic, it is frequently used in high-throughput compound screening. With a 189°C boiling point, it is effectively non-volatile, but small amounts of less than 5 percent are tolerated in conventional electrospray MS. If compound libraries are stored in DMSO, small amounts of the compound will remain in a drug-binding experiment, but providing the concentration is low enough, one might reasonably expect any effect to be minimal. Our experience with DMSO is that its effects on protein structure are both measurable and significant, and it is inconceivable that this would not occasionally alter the outcome of a compound-binding experiment. Worryingly, we find that these conformational alterations occur at any DMSO concentration. It is not difficult to understand how this can happen. In electrospray ionisation (ESI), charged solvent droplets containing the protein of interest are emitted from a spray tip and enter a stream of hot, dry nitrogen gas where desolvation occurs. As the droplet shrinks and surface charge accumulates, a series of Coulombic explosions and
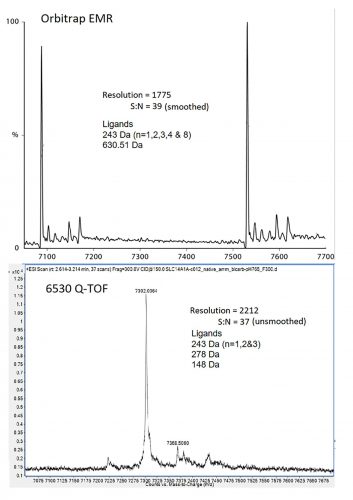
Figure 2: Deconvoluted spectra for integral membrane protein SLC14A1 from Agilent 6530 Q-TOF (top) and Orbitrap EMR (bottom).
further desolvation ultimately leads to ionisation by transfer of the surface charges on to the solvent-free protein. For ESI to work properly, all components in the droplet other than the protein must be volatile. In the case of DMSO, water in the ESI droplet will evaporate at a far greater rate, causing the DMSO to concentrate; so whatever the initial concentration, the final DMSO concentration at the desolvation/ ionisation point will always be significantly higher. The effects of this are dramatic. We tested different concentrations of DMSO on a protein that exists in nature as a 175kDa tetramer with a mean charge radius of 40.7Å and a 350kDa octamer with a mean charge radius of 56.1Å. Even at the lowest concentration, conformational changes equivalent to a 3.2Å reduction in charge radius, or a 19 percent reduction in solvent accessible surface area for the tetramer were observed (Figure 1). Similar DMSO effects have been observed by others.3,7 We do not believe that incorporation of DMSO invalidates native MS screening results, but rather our data suggest that exchanging DMSO for a more volatile solvent such as methanol will drastically improve the results of a native MS compound screen.
Practicalities
 Sample preparation is critically important in any native MS experiment. This involves buffer exchange, usually into a volatile ammonium salt at low molarity pH 6.5 – pH 7.5. This can be carried out offline using three passes through a pre-equilibrated size exclusion spin column; offline by overnight dialysis; or, increasingly, inline and automated by size exclusion LC-MS. Offline automation can be achieved using a Nanomate robot with a microfluidic chip.3 This enables up to 400 MS analyses in a single pass, each acquisition taking just five seconds. A single batch of protein prepared by spin column or dialysis is aliquoted into 96- or 384-well microtitre plates with individual test compounds applied to individual wells. Single samples are usually applied via a static needle drawn by hand and gold-plated for electrical conductivity. Approximately one microlitre of concentrated sample is either drawn into the ESI source by the first stage vacuum or is driven by positive gas pressure. However, in our hands, more dilute sample applied via a syringe pump to a standard nebuliser tip produces equally good spectra.8 Specialised mass spectrometers are not required. All electrospray instruments can operate up to m/z 3,000 and analyse protein-ligand complexes up to 30,000Da. Ion traps have double this m/z range, whereas modern TOFs have m/z ranges of 20,000 or 40,000 and are able to analyse complexes well over 1,000,000Da. High resolution is essential if the bound test compound is to be identified by mass, rather than by a mass-shift associated to its position in the sampling array. Although Orbitrap instruments offer good mass resolution at low mass, this must be somewhat compromised even in instruments specifically designed to operate at high m/z. We have found that equal or better resolution can be achieved using a TOF (Figure 2). MS is an inherently fast technique and recent advances in sample introduction, such as ultrasonic sampling, are expected to establish native MS as a truly high-throughput technique.
Sample preparation is critically important in any native MS experiment. This involves buffer exchange, usually into a volatile ammonium salt at low molarity pH 6.5 – pH 7.5. This can be carried out offline using three passes through a pre-equilibrated size exclusion spin column; offline by overnight dialysis; or, increasingly, inline and automated by size exclusion LC-MS. Offline automation can be achieved using a Nanomate robot with a microfluidic chip.3 This enables up to 400 MS analyses in a single pass, each acquisition taking just five seconds. A single batch of protein prepared by spin column or dialysis is aliquoted into 96- or 384-well microtitre plates with individual test compounds applied to individual wells. Single samples are usually applied via a static needle drawn by hand and gold-plated for electrical conductivity. Approximately one microlitre of concentrated sample is either drawn into the ESI source by the first stage vacuum or is driven by positive gas pressure. However, in our hands, more dilute sample applied via a syringe pump to a standard nebuliser tip produces equally good spectra.8 Specialised mass spectrometers are not required. All electrospray instruments can operate up to m/z 3,000 and analyse protein-ligand complexes up to 30,000Da. Ion traps have double this m/z range, whereas modern TOFs have m/z ranges of 20,000 or 40,000 and are able to analyse complexes well over 1,000,000Da. High resolution is essential if the bound test compound is to be identified by mass, rather than by a mass-shift associated to its position in the sampling array. Although Orbitrap instruments offer good mass resolution at low mass, this must be somewhat compromised even in instruments specifically designed to operate at high m/z. We have found that equal or better resolution can be achieved using a TOF (Figure 2). MS is an inherently fast technique and recent advances in sample introduction, such as ultrasonic sampling, are expected to establish native MS as a truly high-throughput technique.
Biographies
 NICOLA BURGESS-BROWN is the Principal Investigator of the Biotechnology Group at the SGC, responsible for managing all biotech research for the Oxford site. Nicola’s team develops high-throughput screening processes from cloning to expression, purification and mass spectrometry analysis of human proteins for structural and functional studies. Nicola obtained a first class degree in Applied Biochemical Sciences from the University of Ulster in 1997, then worked as a molecular biologist for SmithKline Beecham. She received her PhD in Molecular Microbiology at the University of Nottingham in 2001 and then moved back to industry to work on high-throughput cloning and validation of therapeutic cancer antigens for Oxford Glycosciences and subsequently Celltech R&D.
NICOLA BURGESS-BROWN is the Principal Investigator of the Biotechnology Group at the SGC, responsible for managing all biotech research for the Oxford site. Nicola’s team develops high-throughput screening processes from cloning to expression, purification and mass spectrometry analysis of human proteins for structural and functional studies. Nicola obtained a first class degree in Applied Biochemical Sciences from the University of Ulster in 1997, then worked as a molecular biologist for SmithKline Beecham. She received her PhD in Molecular Microbiology at the University of Nottingham in 2001 and then moved back to industry to work on high-throughput cloning and validation of therapeutic cancer antigens for Oxford Glycosciences and subsequently Celltech R&D.
 ROD CHALK gained his PhD in 1992 at The Liverpool School of Tropical Medicine, working on the innate immune peptides from the mosquito Aedes aegypti. He began using mass spectrometry for bioactive peptide natural product drug discovery at Queens University Belfast in 1996, subsequently working on SNP genotyping and cryodetector mass spectrometry. More recently, Rod has worked in proteomics and biopharmaceuticals using a variety of techniques and instruments. Rod joined the SGC in 2008. His current interests include high-throughput protein characterisation, membrane proteins, quantitative mass spectrometry and the application of native mass spectrometry to structural biology and drug discovery.
ROD CHALK gained his PhD in 1992 at The Liverpool School of Tropical Medicine, working on the innate immune peptides from the mosquito Aedes aegypti. He began using mass spectrometry for bioactive peptide natural product drug discovery at Queens University Belfast in 1996, subsequently working on SNP genotyping and cryodetector mass spectrometry. More recently, Rod has worked in proteomics and biopharmaceuticals using a variety of techniques and instruments. Rod joined the SGC in 2008. His current interests include high-throughput protein characterisation, membrane proteins, quantitative mass spectrometry and the application of native mass spectrometry to structural biology and drug discovery.
References
- Meiby E, et al. Fragment Screening by Weak Affinity Chromatography: Comparison with Established Techniques for Screening against HSP90. Analytical Chemistry, 2013. 85(14): p. 6756-6766.
- Chalk R, et al. Electrospray Surface Charge Describes Protein Molecular Motion. bioRxiv, 2019: p. 571091.
- Goth M, et al. Critical Evaluation of Native Electrospray Ionization Mass Spectrometry for Fragment-Based Screening. ChemMedChem, 2017. 12(15): p. 1201-1211.
- Laganowsky A, et al. Mass spectrometry of intact membrane protein complexes. Nature Protocols, 2013. 8: p. 639.
- Bern M, et al. Parsimonious Charge Deconvolution for Native Mass Spectrometry. Journal of Proteome Research, 2018. 17(3): p. 1216-1226.
- Schiebel J, et al. One Question, Multiple Answers: Biochemical and Biophysical Screening Methods Retrieve Deviating Fragment Hit Lists. ChemMedChem, 2015. 10(9): p. 1511-1521.
- Chan DSH, et al. Insight into Protein Conformation and Subcharging by DMSO from Native Ion Mobility Mass Spectrometry. ChemistrySelect, 2016. 1(18): p. 5686-5690.
- Burgess-Brown NA. Heterologous Gene Expression in E.coli: Methods and Protocols. 2017: Springer New York.
Related topics
Analytical Techniques, Assays, Mass Spectrometry
Related organisations
Biotechnology Group, Structural Genomics Consortium (SGC)
Related people
Nicola Burgess-Brown, Rod Chalk