Screening and beyond in drug discovery
Posted: 14 June 2019 | Sheraz Gul (Head of Drug Discovery at the Fraunhofer Institute for Molecular Biology and Applied Ecology) | No comments yet
Progressing drug discovery projects to a stage that attracts commercial interest is a challenging endeavour, evidenced by the fact that only a small proportion of drug discovery projects are successfully progressed to such a stage. A major cause of this is the lack of comprehensive profiling of compounds prior to initiating translational in-vivo work. This article describes the minimal information that should be acquired as early as possible for a bioactive compound in the drug discovery value chain.

Over the past few decades the reductionist approach has been extensively employed to feed the industry’s small molecule drug pipelines.1 The adoption of this approach was logical at its inception as it enabled the identification of hit compounds that specifically bind and modulate the activity of their respective drug targets. However, this approach yields compounds that are weakly active, lack selectivity and possess sub-optimal properties. These hit compounds therefore require significant optimisation in order to yield a lead compound2 that is potent with optimised selectivity, physicochemical and ADMET properties.3-4
Hit-to-lead
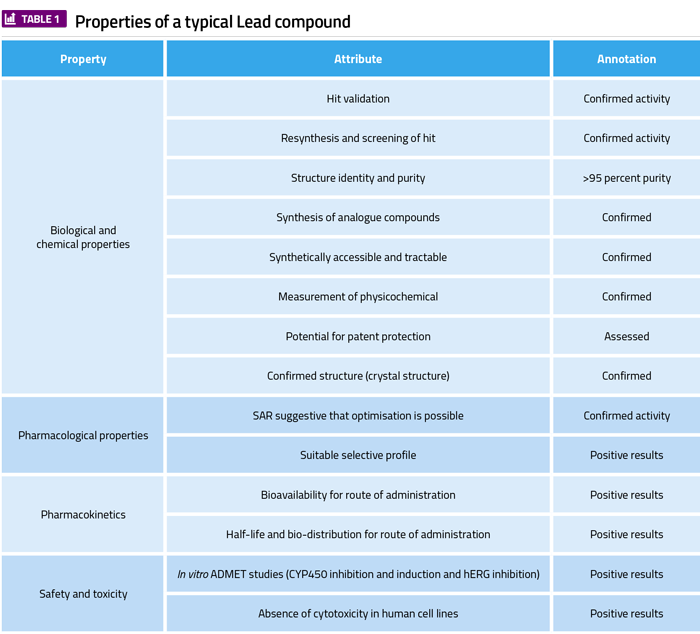
In order to successfully progress compounds to a lead compound, numerous properties must be determined – including adequate biological, chemical, pharmacological and pharmacokinetic characterisation – and each are summarised in Table 1. Most of these studies are in vitro and involve complex processes including cell viability assays. Cell viability relies upon the integrity of the cell membrane, key cellular biochemical reactions and specific cellular markers.5 Knowledge regarding the extent of cytotoxicity that is induced by a compound is a parameter that must be determined prior to progressing it in the drug discovery value chain. A variety of microtitre plate-based cytotoxicity assays in colourimetric, uorometric and luminescence detection technologies are available. The colourimetric-based methods have historically been the most widely employed and are well validated. These include quantification of the following:
- Mitochondrial succinate dehydrogenase activity of cells using the tetrazoles XTT and MTT6
- Extracellular lactate dehydrogenase activity by measuring NADH consumption7
- Acid phosphatase activity as a marker of lysosomal activity8
- Remaining glucose in cell culture medium using a glucose oxidase-peroxidase assay9
- Cell proliferation using crystal violet dye accumulation in the nucleus10
- Lysosomal accumulation of a cationic dye neutral red11
- Protein synthesis using sulforhodamine B binding to proteins.12
Fluorometric-based cytotoxicity assays are also available and include those that quantify the following:
- Lactate dehydrogenase activity using a coupled reaction that results in the conversion of resazurin into resorufin (CytoTox-ONE Assay)13
- Protease activity that is released from cells with a compromised cell membrane and a non-cell permeable fluorogenic peptide substrate (CytoTox-Fluor Cytotoxicity Assay)12
- The activity of two proteases using cell permeable (giving the live cell measurement) and non-cell permeable (giving the dead cell measurement) substrates (MultiTox-Fluor Multiplex Cytotoxicity Assay).14
Luminescence-based assays are also available and include those that quantify the following:
- The activity of a protease that is released from cells that no longer retain an intact cell membrane using a luminogenic peptide substrate (CytoTox-Glo Cytotoxicity Assay)15
- Glutathione-5-transferase activity using a luciferin-derived substrate, the product of the reaction being a substrate of firefly luciferase16
- Intracellular ATP using a luciferase reaction (CellTiter-Glo Luminescent Cell Viability Assay).17
There is also a multiplex assay that uses a fluorescence and luminescence readout essentially the same as the MultiTox-Fluor Multiplex Cytotoxicity Assay mentioned above, except the substrate for dead cells is luminogenic rather than being fluuorogenic.18
Lead-to-candidate
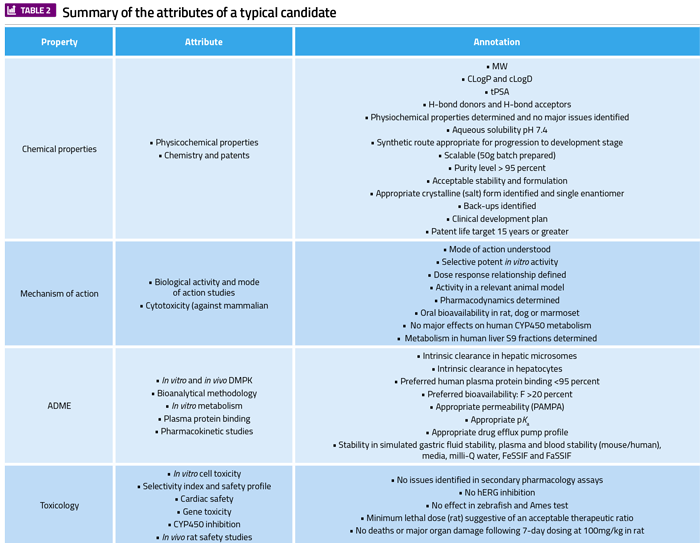
The optimisation of a lead molecule into a candidate is a very challenging, multiparameter process.19-20 It should lead to identification of a molecule that satisfies pre-defined criteria with regard to in vitro and in vivo activity, pharmacokinetics, toxicological and pharmaceutical properties, and complexity of chemical synthesis (Table 2). It will typically have nano-molar potency against its primary biological target and will have undergone significant medicinal chemistry optimisation, comprehensive in vitro selectivity screening, determination of physicochemical and pharmaceutical properties and initial formulation studies, detailed in vitro ADMET studies, mutagenicity testing, detailed secondary pharmacology studies, detailed in vivo pharmacodynamics and pharmacokinetic studies and comprehensive toxicological profiling.
Although drug discovery is fraught with challenges, as highlighted above, the prospect of discovering a new drug is highly rewarding, as it offers the potential for mankind to live healthier and longer lives.
Biography
 SHERAZ GUL is Head of Drug Discovery at the Fraunhofer Institute for Molecular Biology and Applied Ecology in Germany. He is responsible for the management and development of assay development and medium- and high-throughput screening activities for partners across the world. He has 23 years’ experience in both academia (University of London) and industry (GlaxoSmithKline Pharmaceuticals). This has ranged from the detailed study of catalysis by biological catalysts (enzymes and catalytic antibodies) to the design and development of assays for high-throughput screening for the major drug target classes. He is co-author of numerous papers, chapters and the ‘Enzyme Assays: Essential Data’ handbook.
SHERAZ GUL is Head of Drug Discovery at the Fraunhofer Institute for Molecular Biology and Applied Ecology in Germany. He is responsible for the management and development of assay development and medium- and high-throughput screening activities for partners across the world. He has 23 years’ experience in both academia (University of London) and industry (GlaxoSmithKline Pharmaceuticals). This has ranged from the detailed study of catalysis by biological catalysts (enzymes and catalytic antibodies) to the design and development of assays for high-throughput screening for the major drug target classes. He is co-author of numerous papers, chapters and the ‘Enzyme Assays: Essential Data’ handbook.
References
- Blass BE. 2015. Basic Principles of Drug Discovery and Development. Academic Press, USA, pp. 580.
- Thompson AM, et al. 2018. Development of (6 R)-2- Nitro-6-[4-(tri uoromethoxy)phenoxy]-6,7- dihydro-5 H-imidazo[2,1- b][1,3]oxazine (DNDI-8219): A new lead for visceral leishmaniasis. J. Med. Chem. 61: 2329–2352.
- Barberis C, et al. 2017. Discovery of N-substituted 7-azaindoles as Pan-PIM kinase inhibitors-Lead series identification—Part II. Bioorg. Med. Chem. Lett. 27: 4735–4740.
- Shirai J, et al. 2018. Discovery of orally efficacious RORγt inverse agonists, part 1: Identification of novel phenylglycinamides as lead scffolds. Bioorg. Med. Chem. 26: 483–500.
- Méry B, et al. 2017. In vitro cell death determination for drug discovery: A landscape review of real issues. J. Cell Death. 10: 1.
- Berridge MV, et al. 2005. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol. Annu. Rev. 11: 127–152.
- Decker T, et al. 1988. A quick and simple method for the quantification of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumour necrosis factor (TNF) activity. J. Immunol. Methods. 115: 61–69.
- Ivanov DP, et al. 2017. High-throughput spheroid screens using volume, resazurin reduction, and acid phosphatase activity. Methods Mol. Biol. 1601: 43–59.
- Wong AI, Huang D. 2014. Assessment of the degree of interference of polyphenolic compounds on glucose oxidation/peroxidase assay. J. Agric. Food Chem. 62: 4571–4576.
- Feoktistova M, et al. 2016. Crystal violet assay for determining viability of cultured cells. Cold Spring Harb Protoc. 2016: 4.
- Ates G, et al. 2017. Assaying cellular viability using the neutral red uptake assay. Methods Mol. Biol. 1601: 19–26.
- Shaik JS, et al. 2013. Soluble epoxide hydrolase inhibitor trans-4-[4-(3-adamantan-1-yl-ureido)- cyclohexyloxy]-benzoic acid is neuroprotective in rat model of ischemic stroke. Am. J. Physiol. Heart Circ. Physiol. 305: H1605–H1613.
- Shafaie S, et al. 2017. Influence of surface geometry on the culture of human cell lines: A comparative study using at, round-bottom and v-shaped 96 well plates. PLoS One. 12: e0186799.
- Niles AL, et al. 2009. In vitro viability and cytotoxicity testing and same-well multi-parametric combinations for high throughput screening. Curr. Chem. Genomics. 3: 33–41.
- Schorpp K, et al. 2016. A multiplexed high-content screening approach using the chromobody technology to identify cell cycle modulators in living cells. J. Biomol. Screen. 21: 965–977.
- Slim R, et al. 2000. Cellular glutathione status modulates polychlorinated biphenyl-induced stress response and apoptosis in vascular endothelial cells. Toxicol. Appl. Pharmacol. 166: 36–42.
- Lhuissier E, et al. 2017. Identification of an easy to use 3D culture model to investigate invasion and anticancer drug response in chondrosarcomas. BMC Cancer. 17: 490.
- Trumpi K, et al. 2015. Paired image- and FACS-based toxicity assays for high content screening of spheroid-type tumour cell cultures. FEBS Open Bio. 5: 85–90.
- Forkuo GS, et al. 2018. A novel orally available asthma drug candidate that reduces smooth muscle constriction and inflammation by targeting GABAA receptors in the lung. Mol. Pharm. 15: 1766–1777.
- Mikami S, et al. Discovery of clinical candidate N-((1S)-1-(3-Fluoro-4-(trifluoromethoxy)phenyl)- 2-methoxyethyl)-7-methoxy-2-oxo-2,3-dihydropyrido[2,3-b] pyrazine-4(1H)-carboxamide (TAK- 915): A highly potent, selective, and brain-penetrating phosphodiesterase 2A inhibitor for the treatment of cognitive disorders. J. Med. Chem. 60: 7677–7702.
Related topics
Screening
Related organisations
Fraunhofer Institute for Molecular Biology and Applied Ecology
Related people
Sheraz Gul