Off-the-shelf T-cell therapy from gene edited stem cells
Posted: 10 June 2021 | Dr Jo Brewer (Adaptimmune) | No comments yet
Dr Jo Brewer outlines research into allogeneic products by gene editing stem cells, which has the potential to build a broad family of multiple immune cell types, including T-cell therapies.
Breaking the patient manufacturing cycle and enabling immediate access to treatment is an important component in making cell therapies readily available to large number of people with cancer. Generating efficacious allogeneic therapies is therefore a key step in making cell therapy mainstream. The first allogeneic product we hope to have in the clinic will use an engineered T-cell receptor (TCR) targeting MAGE-A4, a TCR and a target that we understand well from clinical trials.
We use human-induced pluripotent stem cells (iPSCs) as the basis for the allogeneic platform because they are amenable to multiple rounds of genetic modification and have a high capacity for proliferation, both of which enable the generation of large iPSC banks. iPSCs are pluripotent, which means they have the potential to generate any human cell type. A single vial from the edited stem cell bank is put through a proprietary differentiation process, which directs the cells down a particular developmental pathway to produce a batch of cells that can treat multiple patients. Each batch of differentiated cells comes from the same starting material in the stem cell bank, which should minimise the variation between batches of the product. In theory, multiple batches of differentiated T cells, from the same iPSC source, could be held at the ready for any eligible patient with cancer.
There are three broad areas to think about when genetically modifying an allogeneic product compared to an autologous one:
- Potency – for our first allogeneic product, we use the same MAGE-A4 targeted TCR currently in clinical trials so that we can gauge the potency in direct comparison with our autologous product. We expect to add further next-generation modifications to improve the product in the future – learning from our patients – just like we do with our autologous platform. Using gene editing in the allogeneic platform will allow us to introduce multiple changes at once.
- Safety – it is vital to make sure that we control the endogenous TCR, or get rid of it altogether, to prevent graft-versus-host disease. The introduced TCR will undergo extensive pre-clinical safety testing to assess its target specificity as we do routinely with our autologous platform.
- Persistence – allogeneic cells are non-self-cells and may be rejected by the patients’ own immune system, but they need to be present long enough to tackle the tumour. To reduce the risk of rejection, we knock-out potentially immunogenic genes. We also knock-in HLA-E so the cells are not targeted for natural killer (NK) cell killing. We know from our autologous products that persistence is likely to be a crucial factor for efficacy, particularly with solid tumours. This is one of the reasons that we are focusing on generating an allogeneic T-cell platform first. T cells should have a longer life span in patients after delivery than some other immune cells and we believe that matters.
We have built a process that mimics early T-cell development in a dish (Figure 1). Normally, T cells develop through the embryo into early life, but we have reduced this to a timescale that is workable for manufacturing. The stem cells start in a completely pluripotent state (shown in the left-hand panel) and will differentiate into any cell type, depending on the cues that they receive. The trick is to get them to differentiate in the way that we want them to — producing alpha beta T cells.
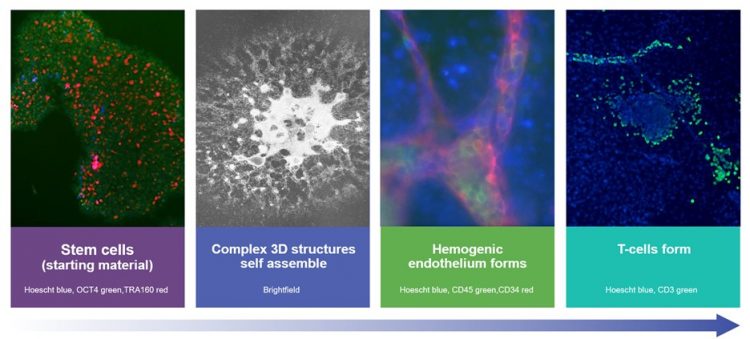
Complex three-dimensional (3D) structures self-assemble in the initial stages of the process (second panel and third panel). This is one of the key differences with our platform and others. These structures support the T-cell differentiation pathway without the need for an accessory feeder or stromal cell line and this is a clear advantage as we move towards good manufacturing practice (GMP) manufacturing. The red tubular structures, in the third image, are stained with an endothelial marker and are like blood vessels in a dish. In key areas these blood vessels are haemogenic, meaning that they generate blood cell precursors inside the vessels, which are stained green. These precursor cells have the potential to generate any blood cells – red blood cells, T cells, B cells, NK cells, macrophages etc. We direct the differentiation of the cells down the T-cell pathway and the last image shows CD3 staining, in green, which confirms that we have made T cells at the end of our process.
The researchers have been working with a dedicated process development team and GMP compatibility has been at the front of their minds as we aim to get these products to the clinic. As such, the research process uses defined media that is serum-free. Human serum is a well-known source of variability and potential viral contamination that can confound GMP production. Similarly, not using feeder cell lines in the process eliminates another potential contaminating element and helps smooth our path towards GMP manufacturing.
We have shown that allogeneic iT cells can kill MAGE-A4 expressing target cells. At the top of Figure 2 is an experiment using control autologous cells targeting MAGE-A4. The black line represents a MAGE-A4 expressing tumour line growing during the assay (GFP positive line, cells counted as green objects). When we add non-transduced T cells, which have not been engineered to kill MAGE-A4, you can see the tumour cells continue to grow – shown by the grey curve increasing over time (overlaying the black curve). When MAGE-A4 targeted autologous T cells are added at 24 hours, shown in purple, the curves start to separate with the MAGE-A4 targeted T-cell cultures heading down which represents target cell killing as the tumour cells are dying.
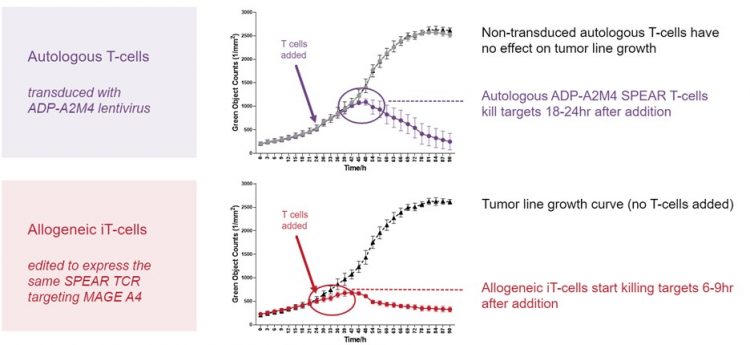
On the bottom panel, the red line shows our allogeneic cells targeting MAGE-A4. The red and black curves clearly separate six to nine hours after the MAGE-A4 targeted cells and control cells are added. The allogeneic cells only kill cells where the MAGE-A4 target is present and the right HLA type is present, just like the autologous cells. The cells that we are generating with this process are potent killers and they keep their TCR specificity, which is important as we move to the clinic.
Using stem cells and gene editing as a base opens up many possibilities for targeting tumours with different receptors and cell types. We believe that the work we are conducting to develop these off-the-shelf therapies will bring these treatments to patients faster and with less complexity in the future.
About the author
Dr Jo Brewer has served as Senior Vice President, Allogeneic Research since December 2019 at Adaptimmune. She leads efforts to generate new, off-the-shelf, cell therapies for people with cancer, as well as work to develop the manufacturing processes for these therapies. Jo has over 19 years of experience in immunotherapy research and was one of the founding scientists at Adaptimmune, working on the early SPEAR T-cell programmes. Jo worked as a Senior Scientist at Avidex (the predecessor company to Adaptimmune), between 2001 and 2006 and remained at Medigene as a Group Leader, following its acquisition of Avidex, until 2008. Jo holds a master’s degree in natural sciences and a PhD in cellular signalling from the University of Cambridge.
Related topics
Immuno-oncology, Immunotherapy, Induced Pluripotent Stem Cells (iPSCs), Oncology, Stem Cells
Related conditions
Cancer