New analytical method provides an insight into additional chromosomes
Posted: 27 February 2018 | Drug Target Review | No comments yet
A new technique promises to identify additional chromosomes involved in carcinogenesis.
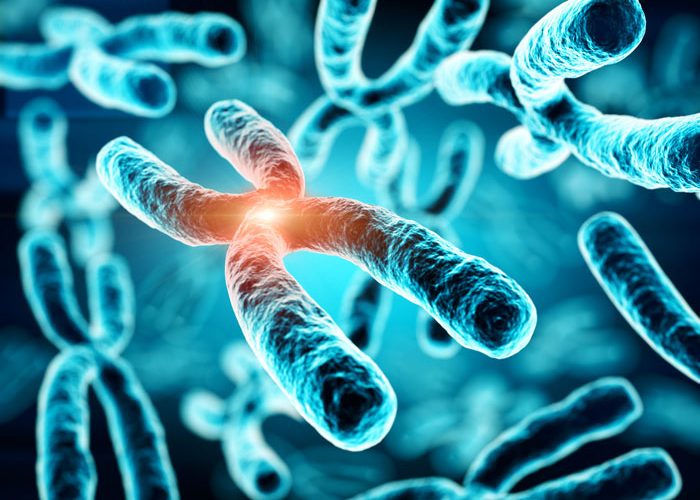
A method for analysing additional chromosomes was proposed by a team of scientists at the Institute of Molecular and Cellular Biology and the Institute of Cytology and Genetics (Siberian branch of Russian Academy of Sciences), NSU Laboratory of Structural, Functional and Comparative Genomics and the University of Belgrade (Serbia) and published in the journal Chromosoma.
Researchers studied the composition of supernumerary human chromosomes and the chromosomes in two mice species. In most animals, the number of chromosomes is stable and one of the most important characteristics of the species. For example, a person has 23 pairs of chromosomes, and a house mouse has 20. However, for some species this number is unstable due to the presence of supernumerary, or additional, chromosomes. Among these species are the common fox, raccoon dog, Siberian roe deer and many species of rodents.
Additional chromosomes are also found in some humans with a probability of 1 in 2,000. Unlike animals in which the presence of additional chromosomes does not appear, 30% of people who are carriers have developmental abnormalities. This is assumed to be because of supernumerary copies of genes contained in these chromosomes.
Alexey Makunin, Candidate of Biological Sciences at the ICG Laboratory of Animal Cytogenetics Laboratory, talked about this work: In our study, small (or thin) additional chromosomes in a human are examined. The principal difference from classical chromosomal pathologies is that an additional copy of a small area is not found on the whole chromosome. The value of the study is that the method we developed identifies which part of the genome is present on the additional chromosome. For example, for Down’s syndrome, the presence of an extra chromosome 21 can be identified using classical methods. We are focusing on more complex cases, more unusual variants of additional chromosomes such as the restructuring in carcinogenesis (formation of malignant tumours). Further development of these tools can be used in both medical and evolutionary genetics.
The study of additional chromosomes is usually complicated by the fact that their DNA is a duplicated portion of the main set of chromosomes. The most frequent additional human microchromosome is a small fragment of chromosome 15. Therefore, when studying a complete genome, these chromosomes merge with the background variability of the main genome. This limitation can be bypassed using microdissection, in which a separate chromosome or even a fragment of one is scratched from the slide with a thin glass needle.
The authors of this study examined additional chromosomes in humans, a yellow-throated mouse (Apodemus flavicollis), and an East Asian mouse (Apodemus peninsulae). Sequencing of individual chromosomes made it possible to ascertain the DNA composition of additional chromosomes and, most importantly, to accurately identify the boundaries of duplicated regions of the genome.
It demonstrated that an additional human microchromosome contains a region of chromosome 15 bounded by highly repetitive DNA centromeres (alpha-satellites), between which an illegal recombination seemed to occur. Additional chromosomes of two mice species consist of a whole set of fragments from the main genome, including genes: 38 in the yellow-throated mouse and 32 in the East Asian mouse. However, the composition of these chromosomes cannot be considered completely random.
In both species, enrichment with genes associated with the cytoskeleton and control of the cell cycle was observed. The kinase gene Vrk1 is common in the additional chromosome, phosphorylating the apoptosis protein p53. Earlier kinase-regulators of the cell cycle were found in additional chromosomes and other mammalian species. According to the authors’ hypothesis, these copies of genes can contribute to the preservation and accumulation of additional chromosomes. The work performed demonstrates that genetic diversity is determined by a complex combination of random and directed processes. Understanding of the non-essential genome elements content provides important insights into genome evolution in general.
Related organisations
Functional and Comparative Genomics, Institute of Molecular and Cellular Biology, NSU Laboratory of Structural, Russian Academy of Sciences, University of Belgrade
Related people
Alexey Makunin