Insights into GPCRs via innovative imaging
Posted: 21 September 2022 | Dr Jonathan Javitch (Columbia University), Ria Kakkad (Drug Target Review) | No comments yet
In this exclusive Q&A, Drug Target Review’s Ria Kakkad spoke with Dr Jonathan Javitch, Professor at Columbia University’s Vagelos College of Physicians and Surgeons, about the cutting-edge imaging method single-molecule fluorescence resonance energy transfer (smFRET), used to investigate G protein-coupled receptors (GPCRs).
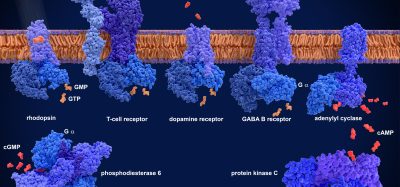
The GPCR family is of high pharmaceutical importance, especially in the drug discovery process, because GPCRs are involved in transmitting external signals into the cell. The GPCR family includes receptors responsible for recognising light, taste, odours, hormones, pain, neurotransmitters and many other stimuli. Therefore, most physiological processes are based on GPCR signalling.
In a recent study carried out at Columbia University Irving Medical Center, US, researchers gained new insights into how GPCRs operate, moving towards a strategy for developing improved drugs with fewer side effects. The study, recently published in Cell,1 involved using an innovative technique called smFRET imaging. The technique captures movements within individual protein systems in unparalleled detail. Moreover, since the method visualises structural changes of single proteins in real time, it enables insights that are obscured by other traditional approaches that average large numbers of proteins in a sample.
With smFRET, the team decided to probe the β-adrenergic receptor – a prototypical GPCR broadly relevant to many different areas of biology. The binding of drugs or endogenous hormones to β-adrenergic receptors or other GCPRs on the cell’s exterior membrane leads to signals on the inside of the cell that are mediated by the activation of G proteins. However, binding of another type of protein, β-arrestins, terminates this signalling and can activate other – desired or undesired – downstream pathways.
By observing the process of β-arrestin activation by a β-adrenergic receptor, the researchers uncovered new details about how β-arrestins interact with and are activated by GPCRs, which require the release of autoinhibition of both proteins.
According to the researchers from the study, their findings could ultimately help to identify improved drugs that, by modulating the binding and/or activation of β-arrestin to GPCRs, affect specific pathways and not others.
Dr Javitch, senior author of the study’s paper, outlines his findings below and provides more detail about the imaging methods used in this research.
Can you briefly outline your research and the imaging methods you used?
The imaging methods use total internal reflection fluorescence microscopy (TIRF) illumination in a prism-based microscope setup, immobilisation of a purified and labelled arrestin sensor that we designed, optimised, purified and labelled in our lab, on a passivated glass surface and imaging of wide field of many individual molecules. Since we used a donor and acceptor dye for labelling, we could capture the distance (and thus conformational change-related dynamics) of the tail of arrestin in the resting state and as it engaged with phosphorylated peptide receptor tail mimics or phosphorylated receptor, made in Robert Lefkowitz’s lab, an author of the study. Using single-molecule FRET as a molecular ruler, we could interrogate mechanisms of arrestin tail release and receptor engagement.
What are some advantages and disadvantages of using smFRET imaging over traditional imaging approaches?
SmFRET allowed us to watch individual proteins at work, allowing us to detect kinetics and mechanisms that are averaged out in typical ensemble studies of huge numbers of proteins.
However, in the current implementation we can only image as rapidly as perhaps 10ms frames. If movements occur on the microsecond time scale, they are averaged out. The work is incredibly technically challenging and slow and required many technical advancements.
What was your most interesting/promising finding?
The C tail of arrestin binds tightly to the N groove in an autoinhibition mechanism. The tail is tightly bound and releases incredibly slowly, too slow for biology. We found that a phosphorylated peptide binds much more slowly than diffusion limited, consistent with the need for arrestin tail release to allow the peptide to bind in the same groove, but the binding is much faster than would be expected if it waited for spontaneous tail release. We think the peptide tickles the extended groove of arrestin to allosterically loosen the tightly bound mid-tail of arrestin to allow the peptide or receptor tail to bind.
We also discovered an unexpected autoinhibitory role for the receptor C tail. Phosphorylation of the tail (even though identical to the free phosphopeptide) was not enough to engage arrestin – agonist was necessary to free the C tail and allow it to engage arrestin and dock into the core of the receptor. We performed cell-based studies to validate an autoinhibitory role of the receptor C tail in living cells.
These studies start to provide insight into the interplay between the ligand bound to the receptor and the pattern of phosphorylation of the receptor and how these are translated in activation of arrestin.
Arrestin not only arrests G protein signalling but also can scaffold other proteins and lead to its own signalling pathways and there is hope of designing ligands that can target one pathway or the other to design better drugs.
What challenges did you face during your study? How did you overcome these?
We had to optimise and go through multiple different tail sensors to find one that worked well. We used new dyes developed over recent years in Scott Blanchard’s lab, the study’s co-senior author, that are brighter, more stable for imaging and longer lived. We used an optimised microscope built in Blanchard’s lab for this type of study as well as new image analysis pipelines and had to use methods for purification of phosphorylated receptor developed in Lefkowitz’s lab. We also used advanced computational methods of analysis performed in Lei Shi’s lab, an author of the study, and we had to develop strategies for live cell studies to translate single molecule findings to living cells. This work took us about eight years from start to finish.
How will innovative imaging strategies like smFRET aid the discovery of novel targets in the future?
We can use these methods with different receptors and ligands to study the mechanisms of activation of both G proteins and arrestin in the hopes of improving drug development by providing unprecedented mechanistic and kinetic insights.
 Dr Jonathan Javitch obtained his BS and MS in Biological Sciences at Stanford University. He completed a joint MD-PhD programme at the Johns Hopkins University School of Medicine. Jonathan did postdoctoral work on the structure of dopamine receptors at Columbia University. He is currently the Lieber Professor of Experimental Therapeutics in Psychiatry and Professor of Molecular Pharmacology and Therapeutics in the Center for Molecular Recognition and in Physiology and Cellular Biophysics at the Columbia University Vagelos College of Physicians and Surgeons, Director of the Lieber Center for Schizophrenia Research and Treatment and Chief of the Division of Molecular Therapeutics at the New York State Psychiatric Institute.
Dr Jonathan Javitch obtained his BS and MS in Biological Sciences at Stanford University. He completed a joint MD-PhD programme at the Johns Hopkins University School of Medicine. Jonathan did postdoctoral work on the structure of dopamine receptors at Columbia University. He is currently the Lieber Professor of Experimental Therapeutics in Psychiatry and Professor of Molecular Pharmacology and Therapeutics in the Center for Molecular Recognition and in Physiology and Cellular Biophysics at the Columbia University Vagelos College of Physicians and Surgeons, Director of the Lieber Center for Schizophrenia Research and Treatment and Chief of the Division of Molecular Therapeutics at the New York State Psychiatric Institute.
Reference
- Asher W, Terry D, Gregorio G, Kahsai A, Borgia A, et al. GPCR-mediated β-arrestin activation deconvoluted with single-molecule precision. Cell. 2022;185(10):1661-1675.e16.
Related topics
GPCRs, Imaging, Microscopy
Related organisations
Columbia University Irving Medical Center