Hit-to-lead in drug discovery
Posted: 4 September 2016 | Douglas B. Kitchen (Albany Molecular Research Inc.), Mark Wolf (Albany Molecular Research Inc.) | No comments yet
Starting in the 1970s, the drug discovery research process changed dramatically. Our understanding of biological targets and pathways grew and the largely empirical and target agnostic in vivo pharmacology approach was replaced with target-centric methods…

Use of in vitro biochemical assays grew in popularity during the 1980s and high-content screening (HTS) of large compound libraries became the norm. One of the advantages of in vivo pharmacology starting points was that in vivo activity was assured. In contrast, HTS of large libraries for single-target activity produces the semblance of success in that large numbers of diverse chemical series are identified as starting points (hits) for further work towards identifying clinical candidates. However, these hits are typically weakly active in a primary screen and do not necessarily possess drug-like characteristics. Therefore, a new process was developed in drug discovery termed hit-to-lead (H2L), or more fully, hit series-to-lead optimisation.
H2L, which explores the chemistry and biology of hits to eliminate dead-end structures and provides improvements to the remaining hit series so that development time and costs are saved, is now a key process in drug discovery organisations. This process explores the chemical space around each hit-series of compounds and narrows it down to more ‘clinic-ready’ lead structures.
The primary goal of H2L research is to identify a few hit series that each demonstrate the promise of producing a drug candidate after focused lead-optimisation efforts. While every programme will have its own idiosyncratic needs, there are a number of general concerns with identifying orally active drug candidates. Measurements such as potency, selectivity, solubility, permeability, metabolic stability, low Cytochrome P450 (CYP) inhibition and good pharmacokinetic (PK) properties tend to apply across most small-molecule discovery programmes.
Biomarkers aren’t just supporting drug discovery – they’re driving it
FREE market report
From smarter trials to faster insights, this report unpacks the science, strategy and real-world impact behind the next generation of precision therapies.
What you’ll unlock:
- How biomarkers are guiding dose selection and early efficacy decisions in complex trials
- Why multi-omics, liquid biopsy and digital tools are redefining the discovery process
- What makes lab data regulatory-ready and why alignment matters from day one
Explore how biomarkers are shaping early drug development
Access the full report – it’s free!
The H2L process has improved over the past decade. At first, compounds were optimised primarily on the basis of their primary biological activity, with little attention given to their fate in later in vivo assays. Screening libraries were often not very ‘drug-like’ and the H2L starting points reflected that fact. Rather than improving success in the clinic, the screening focus had decreased success. In his seminal paper1, Lipinski was among the first to formalise the effects of target-activity focus and the resulting decreased oral bioavailability due to limited aqueous solubility and permeability, giving birth to the ‘rule-of-5’. Lipinski recognised that the invention of larger and larger molecules limited their continued development.
Most H2L processes have increased the use of calculations to confine lipophilicity (logP), molecular weight and a growing number of other molecular descriptors. Even though the pitfalls of chasing potency were clear, many scaffolds continued to fail due to poor drug-like characteristics. Consequently, various new guideposts such as ligand efficiency2 (LE), lipophilic efficiency3 (LipE), and lipophilic ligand efficiency4 (LLE) were introduced. Each of these parameters attempt to penalise compounds that improve potency with unnecessary increases in molecular size and/or lipophilicity.
In the late 1990s and early 2000s, assays were introduced into H2L programmes that attempted to model absorption, distribution, metabolism, excretion and toxicity (ADMET). Due to their relatively low cost, high speed and reliable predictions of in vivo experiments these assays are now used routinely to profile key factors that influence oral bioavailability.
Current approaches in H2L
To illustrate H2L, we will discuss some sample output from an internal HTS. Approximately 200 weak hits were obtained by screening ~110,000 compounds at a single concentration screen, a fairly typical result. Additional concentration response curves showed that 125 hits possessed activities ranging from 62nM – 75M. The relevant question is: what does a good starting point look like? And how do you choose among 100 or more compounds? This selection step is commonly referred to as hit triage5.
The computer-aided drug design (CADD) scientists analysed the confirmed hits and worked with the medicinal chemists to group them into similar scaffolds. Often, the total number of hits from an HTS effort will reduce to groups of 5-10 similar compounds, each containing a common scaffold.
The ‘most active’ compound is not always the best starting point because many parameters need to be optimised to create a successful drug. To help take the focus off any single parameter the ‘Traffic Light’ (TL) approach, as described by Lobell et al. 6, can be used. While the original paper focused on computed parameters, this approach can be extended to include experimental results. The basic premise behind the TL approach is to assign three ranges for each parameter and define them as good (0), warning (+1) and bad scores (+2). The scores for each compound are added across all the scoring parameters to give a final TL score, which can be used for ranking prospects. Since a lower score is more desirable, this is often affectionately termed a ‘golf score.’
Though we have not done so, if desired, the categories can be weighted to reflect their importance. Table 1 below shows the TL analysis of two hits that came out of the example screen. The categories used to help rank hits include calculated parameters such as TPSA and cLogP, experimentally-derived primary assay data, secondary assay data and kinetic solubility. While compound two is the more active of the two compounds, the TL analysis highlights poor ligand efficiency and high cLogP as drawbacks for this starting point. While solubility was not obtained for compound two, it is not expected to exceed the high solubility found in compound one.
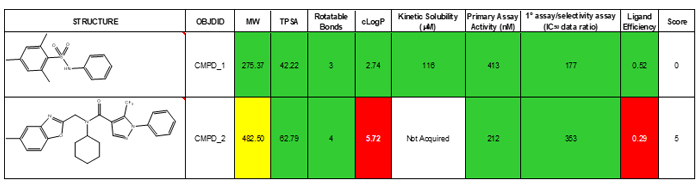
Table 1: Traffic light comparison of two HTS hits.
The flexibility of the TL approach is demonstrated by addition of kinetic solubility and a selectivity assay parameter. Any assay category can be added to the table as H2L proceeds. We have used PAMPA assay data in our TL filter, because the costs and time associated with caco-2 evaluation of hundreds of compounds made it impractical to include. Additionally, the analysis can be performed as aggregate properties of each scaffold. For example, means, medians and standard deviations for each quantity can be summarised.
Once the TL analysis narrows the field to five-10 possible hit series, the rankings of each series can be used to prioritise the resources assigned to their progression. Several critical activities are needed. The activities of the original hits are confirmed independently by purchase or by independent synthesis and rigorous structural analysis. Spurious hits and artifacts are often eliminated by testing in an orthogonal assay that measures binding. Also, mechanism of action studies and experimental assessment of undesirable off-target activities are explored. If possible, new co-crystal structures are obtained. A common tactic in hit triage is to expand potential hits to find similar compounds available for purchase, sometimes referred to as ‘SAR by catalogue.’ Typically, 30-50 compounds are identified, purchased and screened for activity. This approach can be helpful in identifying ‘flat’ SAR where no improvements in activity are achieved, as structures are changed from the original hit or where all changes result in elimination of activity.
H2L begins in earnest as the project team agrees on a set of early ADMET assays to identify weaknesses within a series and to help compare with other potential starting points. Teams commonly monitor solubility, permeability, metabolic stability and CYP inhibition measurements. Early on throughput is often prioritised, but as the programme gets closer to lead optimisation the assays will change. For instance, a single time point for microsomal stability studies may suffice early on, to rank order compounds; later, full determinations of intrinsic clearance in microsomes and/or stability in hepatocytes will be added. In addition to the ADMET assays, proper selectivity assays of related targets need to be evaluated. The team will need to determine whether these counter screens must be routine or if intermittent checks of compounds are sufficient. The key is that the team regularly evaluates their screening funnel to ensure they are collecting the required data to make sound decisions.
With the proper screening funnel in place, it is left to the medicinal chemist to execute a synthetic strategy for each potential hit series to determine the viability of each series. While the primary task of synthesis lies with the medicinal chemist, design and prioritisation of targets is greatly aided using computational resources. These calculations include physicochemical properties such as cLogP, TPSA, rotatable bonds etc., as well as docking models derived from crystal structures of the target, or homologous proteins. The CADD team often provides any secondary activity calculations such as LE, LipE, or LLE to aid the medicinal chemist’s understanding of physicochemical changes that accompany activity gains.
A very important step is to identify existing prior art in order to develop new intellectual property. Early efforts are primarily focused on improving activity at the primary target and finding those changes in structure which eliminate activity. As the potency of a series improves, the chemist considers the physicochemical parameters of new targets as well as their performance in the ADMET and selectivity assays. Even early in the H2L chemistry, the chemist is mindful of creating new intellectual property, as it is important that the drug candidate be protected by patents. Rapid development of diverse synthetic schemes is critical for the exploration of new chemical IP around each series. Frequently, the chemistry team needs to develop multiple convergent synthesis methods in a narrow time window.
Before moving a hit series into lead optimisation, where resources and expenses increase, compounds from that series will need to have demonstrated favourable characteristics as shown in Table 2. During the H2L exploration it is unlikely that all of these qualities will be found in one compound. However, within the series the team is looking for examples that meet the predetermined requirements in these areas. Once those criteria are met, the team can feel confident proceeding into lead optimisation with the series.
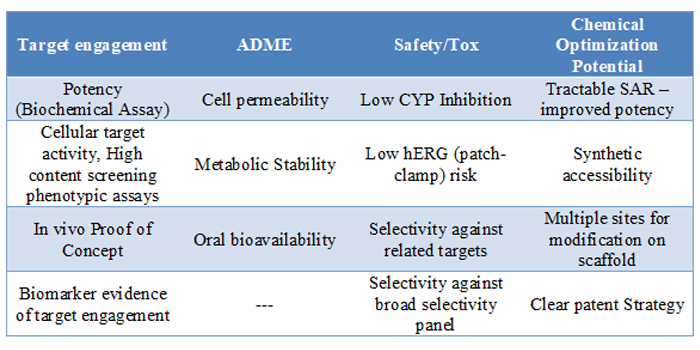
Table 2: Sample evaluation criteria.
The H2L phase of the discovery process is a challenging one. However, with good and regular communication among biology, chemistry, CADD and ADMET team members, the chances of success increase greatly. Tools such as the TL system allow customisation of each HTS effort and are valuable aids in narrowing down the possible starting points.
Biographies
 MARK WOLF has 20 years of drug discovery and development experience at AMRI and has held positions of increasing responsibility during that time. He has led interdisciplinary scientific teams to support large pharma, biotech collaborations and AMRI R&D programmes. His research has spanned several therapeutic areas including oncology, CNS, pain/inflammation and respiratory. Mark has experience establishing practical screening strategies to identify drug candidate compounds with a first-in-class or best-in-class product profile. He has a PhD in organic chemistry.
MARK WOLF has 20 years of drug discovery and development experience at AMRI and has held positions of increasing responsibility during that time. He has led interdisciplinary scientific teams to support large pharma, biotech collaborations and AMRI R&D programmes. His research has spanned several therapeutic areas including oncology, CNS, pain/inflammation and respiratory. Mark has experience establishing practical screening strategies to identify drug candidate compounds with a first-in-class or best-in-class product profile. He has a PhD in organic chemistry.
 DOUG KITCHEN obtained his PhD from Princeton University (Chemistry). After a postdoctoral fellowship at Rutgers University studying biomolecular simulations, he joined Lederle Labs in 1992 in the computer-aided drug discovery group. Doug joined AMRI in 1997 to begin the computational chemistry group. The group has worked on well over 100 projects spanning all aspects of drug discovery. Doug’s interests are in compound library design, structure-guided drug design and lead optimisation. He is a co-inventor on more than 10 issued patents and author on more than forty refereed articles and book chapters.
DOUG KITCHEN obtained his PhD from Princeton University (Chemistry). After a postdoctoral fellowship at Rutgers University studying biomolecular simulations, he joined Lederle Labs in 1992 in the computer-aided drug discovery group. Doug joined AMRI in 1997 to begin the computational chemistry group. The group has worked on well over 100 projects spanning all aspects of drug discovery. Doug’s interests are in compound library design, structure-guided drug design and lead optimisation. He is a co-inventor on more than 10 issued patents and author on more than forty refereed articles and book chapters.
References
- Lipinski CA, Lombardo F, Dominy BW, Feeny PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997; 23: 3-25
- Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Disc Today. 2004; 9 (10): 430-431
- Ryckmans T, Edwards MP, Horne VA, Correia AM, Owen DR, Thompson LR, Tran I, Tutt MF, Young T. Rapid assessment of a novel series of selective CB2 agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis. Bioorg Med Chem Letters. 2009; 19: 4406-4409
- Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Disc. 2007; 6:881-890
- Duffy BC, Zhu L, Decornez H, Kitchen DB. Early phase drug discovery: Cheminformatics and computational techniques in identifying lead series. Bioorg Med Chem. 2012, 20: 5324-5342
- Lobell M, Hendrix M, Hinzen B, Keldenich J, Meier H, Schmeck C, Schohe-Loop R, Wunberg T, Hillisch A. In Silico ADMET Traffic Lights as a Tool for the Prioritization of HTS Hits. Chem Med Chem. 2006; 1: 1229-1236
Related topics
Assays, Biomarkers, Drug Discovery, High-Throughput Screening (HTS), Hit-to-Lead, In Vivo