New frontiers in G protein-coupled receptor drug discovery
Posted: 10 December 2018 | Christopher J. Langmead | No comments yet
G protein-coupled receptors (GPCRs) are seven transmembrane spanning proteins that mediate the physiological responses to a broad array of stimuli, including photons, biogenic amines, peptides and large proteins. They represent the target of approximately one-third of all approved drugs,1 yet paradoxically remain a relatively under-exploited protein class.
Only 134 receptors out of approximately 360 GPCRs in the human genome are currently targets of drugs approved in the USA or European Union.1 This analysis also excludes over 400 olfactory, taste and visual GPCRs, some of which may also represent novel discovery opportunities.2,3
Historically, GPCRs have been targeted for drug discovery by designing compounds that interact with the orthosteric-binding site; ie, that utilised by the endogenous ligand(s) for the receptor. This is probably best exemplified by the development of the blockbuster histamine H2 receptor antagonist, cimetidine (Tagamet), an analogue of the endogenous agonist, histamine, for the treatment of gastric acid secretion.4 Similar approaches have led to many successful drugs acting on GPCRs, yet challenges remain when targeting the wider receptor family. Whilst this partly reflects a paucity of biological knowledge for many GPCRs, efforts are also hampered by limited chemical tractability. This can manifest as a total absence of tractable small molecule chemistry, or be more nuanced; for example, by compounds lacking appropriate selectivity.
Two growing and complementary areas of research are seeking to address these challenges; namely, the development of structural biology approaches to visualise GPCRs at a near-atomic level and targeting allosteric binding sites with novel chemistry to improve tractability.
A new age of GPCR structural biology
Medicinal chemistry for GPCR projects has been predominantly empirical and driven by an understanding of ligand pharmacophore, rather than the structure-based approach that is commonplace for chemistry programmes for soluble proteins such as kinases, phosphatases and proteases.5,6 There is a comparative paucity of high-resolution structural information for GPCRs; which, as membrane proteins, are inherently flexible and able to adopt multiple conformations. This ability is key to their function as transducers of extracellular stimuli to intracellular effector proteins, but severely limits structural approaches that rely on isolating a single conformation of a protein.
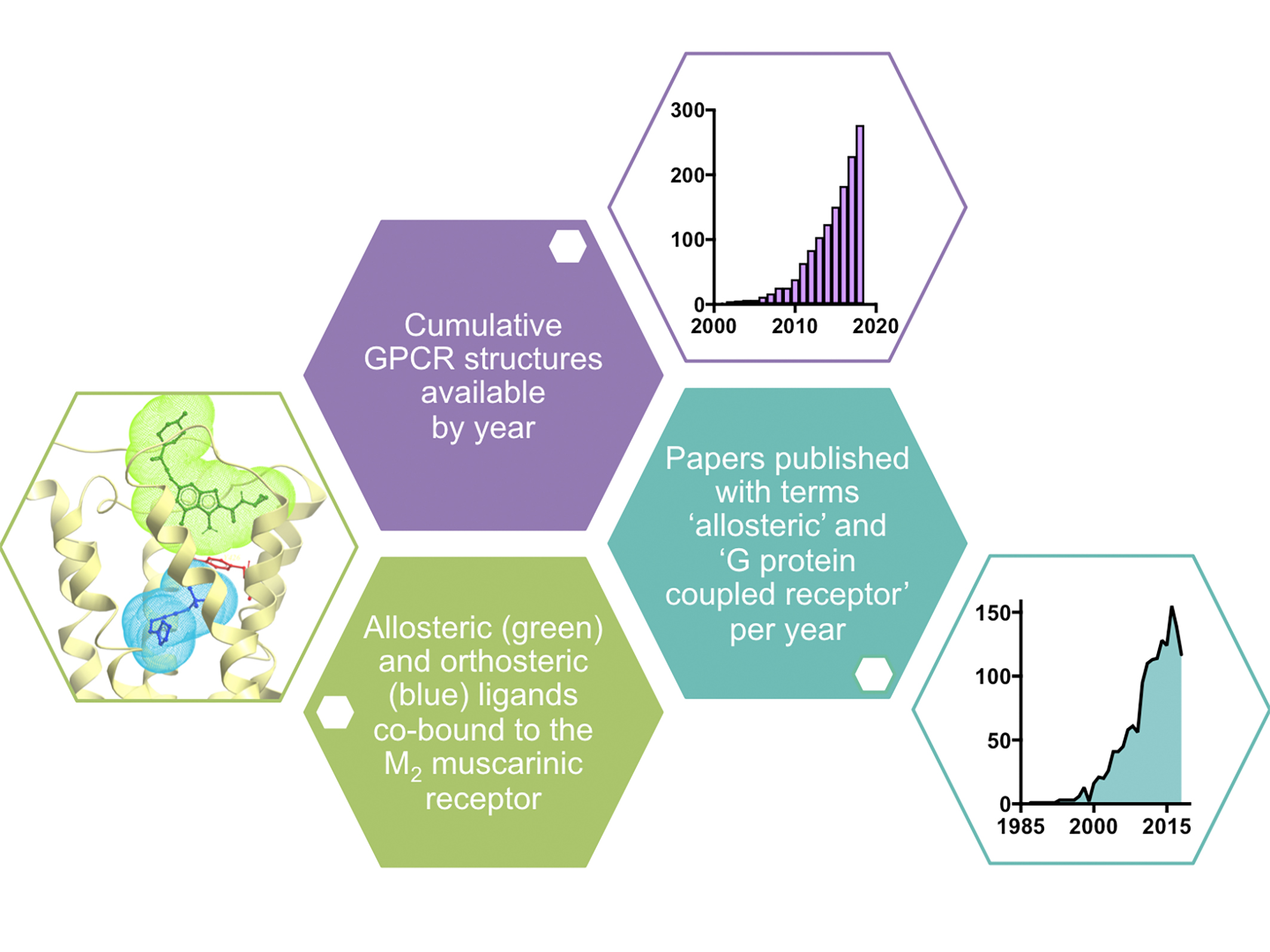
Figure 1. Example of crystal structure of the M2 muscarinic receptor with co-bound orthosteric and allosteric ligands33 and data showing the growing number of GPCR structures24 (as of September 2018) and publications on allosteric modulation of GPCRs by year (taken from Scopus).
However, since the first X-ray crystal structure of a GPCR (rhodopsin) was solved in 2000,7 there has been much progress in techniques available to resolve the structures of GPCRs, and consequently the past five years has seen an exponential growth in the number of new structures deposited into the Protein Data Bank (PDB; Figure 1). Early structures were solved using combinations of approaches to isolate, stabilise and crystallise GPCR proteins, including complexes with antibody fragments,8 engineering fusion proteins,9 introduction of conformationally stabilising mutations10 and the development of a ‘lipidic-cubic phase’ to crystallise GPCRs in a lipid environment that mimics the cell membrane.11
These approaches represented huge breakthroughs, giving researchers insights into drug-receptor interactions at the atomic level. However, these early structures represented snapshots of GPCRs in their inactive, inhibitor (or antagonist)-bound states. Not until 2011 were the first agonist-bound, active state structures solved for the β2-adrenoceptor,12,13 revealing the extent of the conformational changes that take place upon receptor activation and binding of the cognate G proteins to the intracellular face of the receptor. This represented a new frontier in GPCR biology, yielding not only an appreciation of receptor dynamics, but also the binding modes and mechanisms of GPCR agonists.12-15 In the past two years advances in the technology of cryo-electron microscopy have enabled this approach to be used to determine the structures of active-state complexes of GPCRs (including previously unseen extracellular N-terminal regions), ligands and heterotrimeric G proteins.16-20 These structures are providing greater understanding as to how GPCRs function as part of macromolecular complexes, as well as insights into active confirmations engendered by ligands with different pharmacology at the same receptor.21
The key marker for the impact of GPCR structures will be how much of a difference they make to medicinal chemistry efforts targeting this receptor family. Whilst the state-of-the-art is not at the level enjoyed by those working with soluble proteins, there are several examples of GPCR structures being used to identify, design and optimise compounds; notably for the adenosine A2A22 and dopamine D4 receptors.23 Surprisingly, the average resolution of the GPCR structures solved to date is below three angstroms (2.9 ± 0.6Å, n = 27024). Furthermore, the availability of good quality datasets has permitted in silico screening of virtual chemical databases to identify new chemotypes; improved the quality of homology models for GPCRs closely related to solved protein structures; and enabled broader chemogenomic approaches to identify novel ligands for orphan receptors.25
More specifically, structural information has now been used to answer receptor-specific questions, such as understanding the molecular basis for biased agonism at the 5-HT2B receptor26 and why Frizzled receptors are so poorly tractable to small molecule drug discovery (the transmembrane domain binding pocket for the Frizzled-4 receptor is almost impossible for compounds to access).27
The increased availability of structural data (Figure 1), the increased availability of the necessary biochemistry to enable structure determination, and the improvements in associated imaging technology should all bear significant fruit in the coming decade and open up the wider GPCR superfamily to tractable, structure-based drug discovery.
Exploiting allosteric modulation to improve tractability
Alongside the rise in structural data has come an interest in targeting allosteric binding sites on GPCRs. These sites on a receptor are topographically distinct from the orthosteric-binding site and, as such, offer the potential to engender greater levels of receptor selectivity than might be afforded by targeting evolutionarily conserved endogenous agonist binding sites. By binding to a second site on the receptor, allosteric modulators can change the conformation of a GPCR to make it more or less sensitive to the endogenous agonist. The concept of allosteric interactions was described over fifty years ago28 and extended to GPCRs in the late 1970s with agents such as gallamine acting at muscarinic acetylcholine receptors.29
However, it took the advent of functional high-throughput screening assays (which did not distinguish between compounds acting at allosteric versus orthosteric binding sites) in the early 2000s to bring this class of compounds to the attention of the drug discovery industry. The rise in their popularity is evident by the dramatic increase in the number of publications containing the terms “G protein-coupled receptor” and “allosteric” (Figure 1) in the past decade.
Their most attractive trait is that allosteric ligands often exhibit degrees of selectivity not achievable by targeting the orthosteric-binding site. This is perhaps most evident amongst the aforementioned muscarinic acetylcholine receptor family, where much effort has been made to develop subtype-selective agents, particularly for the muscarinic M1 and M4 receptor subtypes, with only modest success.30 However, highly subtype-selective positive allosteric modulators of both receptor subtypes are in late-stage pre-clinical and early clinical development.31 These agents not only possess improved subtype selectivity, but their allosteric mechanism of action preserves the spatial and temporal nature of target receptor signalling and engenders an effect that is saturable, such that on-target side effects can be mitigated.32 It is this same saturable allosteric effect that renders benzodiazepine drugs relatively safe in overdose compared to barbiturates, albeit at a different target class of proteins (GABAA receptors, which are ligand-gated ion channels).32 X-ray crystallography studies have also led to mechanistic understanding of allosteric modulation, with the first structure of an allosteric ternary complex of receptor and two co-bound ligands solved for the muscarinic M2 receptor with the orthosteric agonist, iperoxo, and the positive allosteric modulator, LY2119620 (Figure 1).33
The most successfully exploited GPCRs by allosteric modulation in drug discovery are the Class C family, which includes the family of metabotropic glutamate (mGlu) receptors and the calcium-sensing receptor. Three of the four approved GPCR drugs with an allosteric mechanism of action act via the calcium-sensing receptor (cinacalcet, evocalcet and etelcalcitide) and are approved for various forms of secondary hyperparathyroidism, whereas positive and negative allosteric modulators acting at the mGlu2, mGlu4 and mGlu5 receptor subtypes are in active clinical development. A full list of GPCR allosteric modulator drugs that are either approved or in active clinical development is shown in Table 1 (this excludes compounds for which development programmes have been terminated). Interestingly, this list includes the first example of a GPCR allosteric antibody, namacizumab, which is a negative modulator of the cannabinoid CB1 receptor for the treatment of non-alcoholic fatty liver disease (NAFLD).
It is clear from the data presented above that the growth in the clinical development pipeline of GPCR allosteric ligands – supported by a broad range of pre-clinical development programmes and coupled with the rise of structural insights into GPCRs – is yielding a significant impact in the field. These approaches are enabling opportunities for previously ‘hard to target’ GPCRs, opening up a broad array of untapped potential across this target class.
Table 1. Allosteric modulators of GPCRs either approved or in active clinical development (as of September 2018; data from Cortellis Competitive Intelligence database search*)
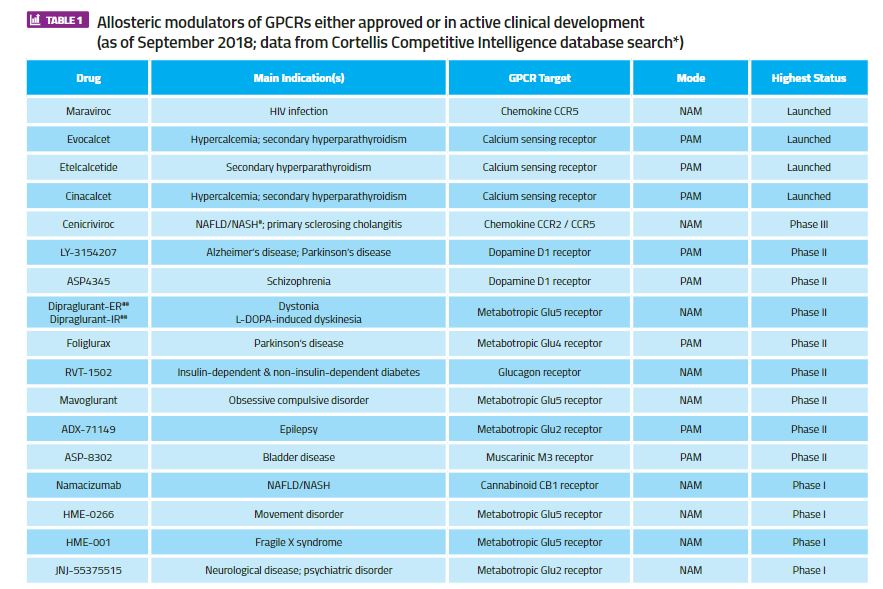
#NAFLD = non-alcoholic fatty liver disease; NASH = non-alcoholic steatohepatitis*Search in Cortellis Competitive Intelligence for drugs with ‘Any Action (G protein-coupled receptor modulator) AND Any Text (“allosteric”)’ filtered for ‘Highest Status’ = ‘Launched; Phase 1 Clinical; Phase 2 Clinical; Phase 3 Clinical’, with minor manual curation for missing, duplicated or incorrectly annotated entries).
**PAM = positive allosteric modulator; NAM = negative allosteric modulator
##IR = immediate release formulation; ER = extended release formulation
Biography
Christopher J. Langmead is Head of the Servier Drug Discovery Program at the Monash Institute of Pharmaceutical Sciences (MIPS), a collaborative programme targeting GPCRs across a range of indications. Prior to this, he was Head of Pharmacology at Heptares Therapeutics and a neuroscience researcher at GlaxoSmithKline. He has a degree and PhD in pharmacology and is a Fellow of the British Pharmacological Society.
References
- Sriram K, Insel PA. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Molecular Pharmacology. 2018;93(4):251-258.
- An SS, Liggett S. Taste and smell GPCRs in the lung: Evidence for a previously unrecognized widespread chemosensory system. Cellular Signalling. 2018;41:82-88.
- Ferrer I, Garcia-Esparcia P, Carmona M, Carro E, Aronica E, Kovacs G et al. Olfactory Receptors in Non-Chemosensory Organs: The Nervous System in Health and Disease. Frontiers in Aging Neuroscience. 2016;8.
- Brimblecombe RW, Duncan WA, Durant GJ, Emmett JC, Ganellin CR, Leslie GB et al. Characterization and development of cimetidine as a histamine H2-receptor antagonist. Gastroenterology. 1978;74(2 Pt 2):339-47.
- Noble M. Protein Kinase Inhibitors: Insights into Drug Design from Structure. Science. 2004;303(5665):1800-1805.
- Murray C, Blundell T. Structural biology in fragment-based drug design. Current Opinion in Structural Biology. 2010;20(4):497-507.
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289(5480):739-45.
- Rasmussen S, Choi H, Rosenbaum D, Kobilka T, Thian F, Edwards P, et al. Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature. 2007;450(7168):383-387.
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007318(5854):1266-73.
- Warne T, Serrano-Vega M, Baker J, Moukhametzianov R, Edwards P, Henderson R et al. Structure of a β1-adrenergic G-protein-coupled receptor. Nature. 2008;454(7203):486-491.
- Caffrey M. Crystallizing Membrane Proteins for Structure Determination: Use of Lipidic Mesophases. Annual Review of Biophysics. 2009;38(1):29-51.
- Rasmussen S, DeVree B, Zou Y, Kruse A, Chung K, Kobilka T, et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nature. 2011;477(7366):549-555.
- Rasmussen S, Choi H, Fung J, Pardon E, Casarosa P, Chae P, et al. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature. 2011;469(7329):175-180.
- Nygaard R, Zou Y, Dror R, Mildorf T, Arlow D, Manglik A, et al. The Dynamic Process of β2-Adrenergic Receptor Activation. Cell. 2013;152(3):532-542.
- Bennett K, Tehan B, Lebon G, Tate C, Weir M, Marshall F, et al. Pharmacology and Structure of Isolated Conformations of the Adenosine A2A Receptor Define Ligand Efficacy. Molecular Pharmacology. 2013;83(5):949-958.
- Liang Y, Khoshouei M, Radjainia M, Zhang Y, Glukhova A, Tarrasch J, et al. Phase-plate cryo-EM structure of a class B GPCR–G-protein complex. Nature. 2017;546(7656):118-123.
- Zhang Y, Sun B, Feng D, Hu H, Chu M, Qu Q, et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature. 2017;546(7657):248-253.
- Koehl A, Hu H, Maeda S, Zhang Y, Qu Q, Paggi JM, et al. Structure of the μ-opioid receptor-Gi protein complex. Nature. 2018;558(7711):547-52
- Draper-Joyce CJ, Khoshouei M, Thal DM, Liang YL, Nguyen ATN, Furness SGB, et al. Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature. 2018;558(7711):559-563.
- García-Nafría J, Nehmé R, Edwards P, Tate C. Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go. 2018;558(7711):620-623.
- Liang YL, Khoshouei M, Glukhova A, Furness SGB, Zhao P, Clydesdale L, et al. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex. Nature. 2018;555(7694):121-125.
- Congreve M, Andrews S, Doré A, Hollenstein K, Hurrell E, Langmead C, et al. Discovery of 1,2,4-Triazine Derivatives as Adenosine A2A Antagonists using Structure Based Drug Design. Journal of Medicinal Chemistry. 2012;55(5):1898-1903.
- Wang S, Wacker D, Levit A, Che T, Betz R, McCorvy J, et al. D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science. 2017;358(6361):381-386.
- https://zhanglab.ccmb.med.umich.edu/GPCR-EXP/
- Roth BL, Irwin JJ, Shoichet BK. Discovery of new GPCR ligands to illuminate new biology. Nature Chemical Biology. 2017;13:1143–1151.
- McCorvy JD, Wacker D, Wang S, Agegnehu B, Liu J, Lansu K, et al. Structural determinants of 5-HT2B receptor activation and biased agonism. Nat Struct Mol Biol. 2018;25(9):787-796.
- Yang S, Wu Y, Xu TH, de Waal PW, He Y, Pu M, et al. Crystal structure of the Frizzled 4 receptor in a ligand-free state. Nature. 2018 Aug;560(7720):666-670.
- Monod J, Wyman J, Changeux J-P. One the nature of allosteric transitions: a plausible model. J Mol Biol. 1965;12:88-118.
- Clark AL, Mitchelson F. The inhibitory effect of gallamine on muscarinic receptors. Br J Pharmacol. 1976;58(3): 323–331.
- Langmead CJ, Watson JM, Reavill C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther. 2008;117(2):232-43.
- https://www.aspet.org/aspet/news/news/2018/01/05/from-bench-to-bedside-how-allosteric-modulators-of-muscarinic-receptors-found-their-way-into-the-clinic
- Christopoulos A, Changeux JP, Catterall WA, Fabbro D, Burris TP, Cidlowski JA, et al. International Union of Basic and Clinical Pharmacology. XC. multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev. 2014;66(4):918-47.
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013 Dec 5;504(7478):101-6.
Related topics
GPCRs, Molecular Biology, Molecular Targets, Protein, Protein Expression
Related organisations
Monash Institute of Pharmaceutical Sciences (MIPS)