Developability assessment of therapeutic antibodies
Posted: 8 March 2017 | David Matthews (MRC Technology), Lone Friis (MRC Technology) | No comments yet
Developability assessment is a broad term covering the evaluation of potential therapeutic candidates’ drug-like properties, manufacturability and safety profile. It is important to carry out this assessment as early as possible in the pre-clinical stage development to select the candidate with the most stable profile. This can minimise the risks of costly late-stage failures for industry and hopefully deliver safer drugs to patients…

State-of-the-art biophysical characterisation tools and methods capable of predicting late-stage behaviour of potential therapeutic candidates are becoming increasingly available. Here we describe a selected range of commonly used techniques to characterise the biophysical stability properties of therapeutic antibodies in early-stage development. An example of relevant parameters to consider is shown in Figure 1.
Purity and fragmentation
Many biophysical techniques are influenced by the presence of impurities or process-related aggregation/fragmentation and consequently it is important to carry out the analytical testing with pure homogenous protein in order evaluate the data appropriately. Typically, both a capture and a polishing step are necessary to achieve this. SDS-PAGE is commonly used to assess purity but lacks the sensitivity and accuracy of SDS capillary electrophoresis (CE-SDS), size exclusion high performance liquid chromatography (SEC-HPLC) and liquid chromatography-mass spectrometry (LC-MS). Highly stable antibodies tend to have good purity profiles and therefore any presence of fragmentation, aggregation or co-purified impurities can give an early warning sign of potentially poor biophysical characteristics.
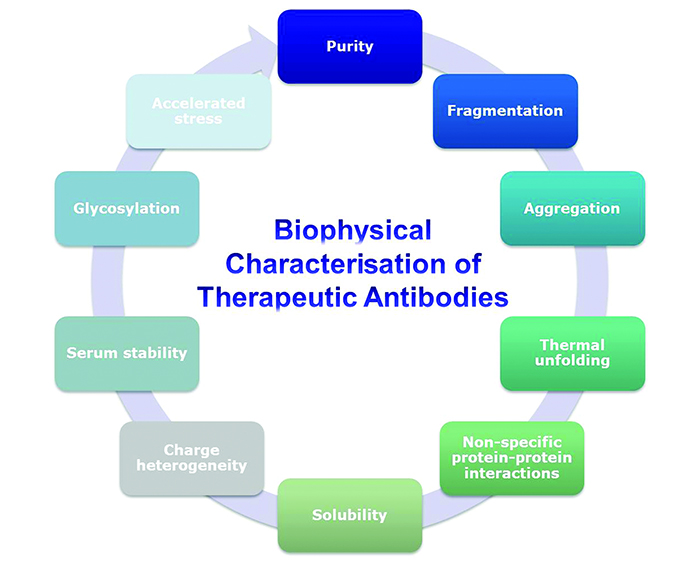
Figure 1: Biophysical properties of antibodies
Aggregation
Aggregation is one of the most critical factors to assess for biologics and many of the biophysical properties shown in Figure 1 can lead to aggregation. Propensity to aggregate can cause several issues in relation to manufacturing, shelf life, patient administration and immunogenicity.1 Widely accepted and complementary tools to assess aggregation are Dynamic light scattering (DLS) and size-exclusion chromatography in line with multi-angle light scattering (SEC-MALS).2
In SEC-MALS, the protein species in a solution are separated according to molecular size on a SEC column prior to passing through, first a UV detector and a refractive index (RI) concentration detector, and finally a MALS detector (three detector analysis). This provides both quantitative and qualitative information about the relative distribution of monomer,
Purity and fragmentation
Many biophysical techniques are influenced by the presence of impurities or process-related aggregation/fragmentation and consequently it is important to carry out the analytical testing with pure homogenous protein in order evaluate the data appropriately. Typically, both a capture and a polishing step are necessary to achieve this. SDS-PAGE is commonly used to assess purity but lacks the sensitivity and accuracy of SDS capillary electrophoresis (CE-SDS), size exclusion high performance liquid chromatography (SEC-HPLC) and liquid chromatography-mass spectrometry (LC-MS). Highly stable antibodies tend to have good purity profiles and therefore any presence of fragmentation, aggregation or co-purified impurities can give an early warning sign of potentially poor biophysical characteristics.
Aggregation
Aggregation is one of the most critical factors to assess for biologics and many of the biophysical properties shown in Figure 1 can lead to aggregation. Propensity to aggregate can cause several issues in relation to manufacturing, shelf life, patient administration and immunogenicity.1 Widely accepted and complementary tools to assess aggregation are Dynamic light scattering (DLS) and size-exclusion chromatography in line with multi-angle light scattering (SEC-MALS).2
In SEC-MALS, the protein species in a solution are separated according to molecular size on a SEC column prior to passing through, first a UV detector and a refractive index (RI) concentration detector, and finally a MALS detector (three detector analysis). This provides both quantitative and qualitative information about the relative distribution of monomer, dimer, trimer and larger aggregate species within the sample. Typically, monomer, dimer and to extend trimer peaks are mono-disperse and will show expected molar masses. By contrast, larger aggregates are polydisperse due to their non-homogenous nature and show increasing molecular weight across the eluting peak. Combining SEC with MALS enables the absolute molar mass measurement of the eluting species independently of retention time; therefore atypical retention on the SEC column does not interfere with the analysis. MALS also has the advantage of a very high sensitivity enabling the detection of low-level large aggregates. The drawbacks of this method are related to the SEC properties such as limited molecular weight separation range, potential adsorption of aggregates to the resin thereby omitting the detection of these, and the effect that dilution and shear forces can have on the equilibrium of the oligomers in solution.
A complementary technique, dynamic light scattering (DLS), involves the analysis of the unseparated sample and as a result does not suffer the drawbacks described above for SEC-MALS. DLS measures fluctuations in scattered laser light intensity caused by the Brownian motion of molecules in solution. This leads to overall sample information regarding size distribution by mass and intensity (typically low intensity/high mass signal for monomer content and vice versa for aggregates), sample polydispersity, hydrodynamic radius and average molecular weight. In contrast to SEC-MALS, DLS is compatible with most buffers, which makes it a suitable technique for examining buffer and pH effects on aggregation in a high throughput manner. Nevertheless, due to the lack of separation of the monomer and oligomer species the method has poor resolution and only yields qualitative data.
Thermal unfolding
Instability caused by unfolding of the antibody can cause irreversible aggregation and loss of activity. Such instability can be uncovered by high-throughput methods with low sample requirements, such as thermal shift by differential scanning fluorimetry (DSF), which involves thermal denaturation analysis of the whole IgG molecule, and a complementary thermal stability binding assay, which focuses on the retention of binding capability after incubation at a range of increasing temperatures (Figure 2b). DSF is carried out with a fluorescent dye (eg, Sypro Orange), which is highly fluorescent in a hydrophobic environment.
As the protein gradually unfolds upon increasing temperature more hydrophobic pockets become accessible for the dye and the signal increases until the protein is completely denatured. The melting temperature (Tm) of the protein is equal to the half maximal point of the fluorescence peak and is commonly used to describe the overall conformational stability of an antibody (Figure 2a).3 Whereas DSF requires purified protein, the thermostability binding assay has the advantage that it can be carried out with crude supernatants by either ELISA or flow cytometry and therefore eliminate particular unstable candidates at the very early selection stages.
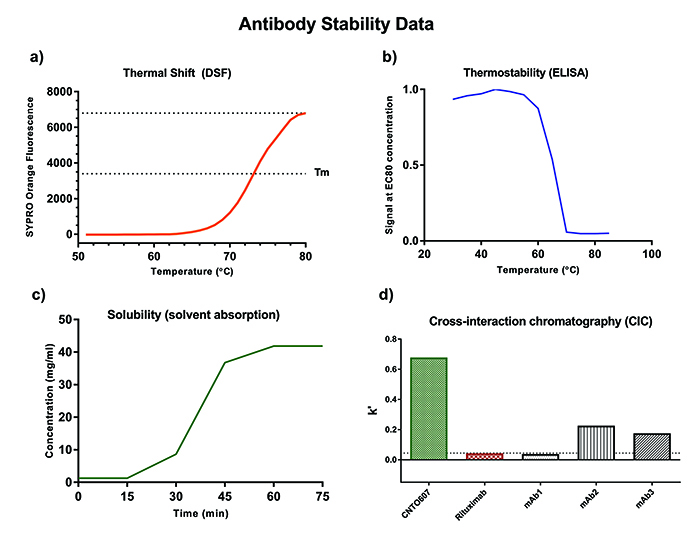
Figure 2: Antibody stability data
Non-specific protein-protein interactions
Non-specific protein-protein interactions can negatively impact solubility, viscosity, and aggregation propensity of an antibody at high concentrations and thus is an important factor to consider in relation to manufacturing, where high concentrations are achieved during purification, and in delivery formulations requiring low volumes with high drug concentration. In addition, pharmacokinetics properties can also be affected.4 A range of different methods have been developed to assess the tendency of an antibody to engage in non-specific protein-protein interactions through interactions with either self:
- Self-interaction chromatography (SIC)5
- Affinity-capture self-interaction nanoparticle spectroscopy (AC-SINS)6
- Clone self-interaction biolayer interferometry (CSI-BLI)7 or with a polyclonal IgG mixture
- Cross-interaction chromatography (CIC)8 (Figure 2d).
Some of these methods have limited reproducibility and batch-to-batch variation due to the need for column/particle preparations. A more recent alternative to the above techniques is standup monolayer adsorption chromatography (SMAC), which is a SEC column-based approach. The particular resin is a hydrophobic standup monolayer with terminal hydrophilic groups that mimic the exterior of a protein and causes delayed retention of “sticky” antibodies. This method has shown good correlation with CIC and benefits from a commercially available column produced under controlled conditions.9
Solubility
Any therapeutic drug must have good solubility in order to administer to the patient in realistic volumes. For subcutaneous delivery, concentrations of 50-100mg/ml or higher must be achieved due to dosing volumes down to 0.5ml. Intravenous delivery enables injection of larger volumes, so lower concentrations are typically needed.10 Poor solubility can be related to hydrophobicity and tendency to aggregate. Direct measurement of solubility requires significant amounts of purified antibody, which is rarely available at the early candidate screening stage. Several surrogate methods requiring small amounts of antibody have been developed to show some correlation with solubility.
Increased non-specific protein-protein interactions often lead to poor solubility; hence all of the assays described in the above section can give some indication of relative solubility in ranking experiments.11 Other assays include second virial coefficient (B22) determination by DLS and PEG/ammonium sulphate-based precipitation assays.12,13 Care must be taken not to over-interpret results from these assays and a direct measurement of solubility should be carried out as soon as possible in the lead candidate selection phase.
Direct measurements of solubility are typically carried out by centrifugal ultrafiltration in spin concentrators, followed by quantification of protein concentration by UV-spectrometry. This method subjects the antibody to shearing forces and pressure-induced stress, which can falsely impact the results. A gentler alternative to ultrafiltration is use of static solvent absorption concentrators, where a membrane of selective permeability is backed by a
pressure resistant absorbent pad that pulls solvent and salts through the membrane and thereby concentrates the sample (Figure 2c).
Charge heterogeneity
Charge heterogeneity is linked to post-translational modifications of charged residues in the primary amino acid sequence, such as deamidation, isomerisation, succinimide formation, oxidation, sialylation, N-terminal pyroglutamic acid or C-terminal lysine (Lys) clipping, which create a number of acidic and basic isoforms.14 Changes in the manufacturing process can alter the charge heterogeneity profile of an antibody and impact the antibody’s biological activity and stability. Therefore it is important to identify candidates that have a stable charge heterogeneity profile and ideally few major isoforms. This can also provide a fingerprint for easy analysis of batch-to-batch variation.
The analysis of charge variants can be carried out by a variety of capillary electrophoresis (CE) based methods and characterised in detail in combination with mass spectrometry. CIEF determines the isoelectric point (pI) of an antibody preparation and is governed by the heterogeneity of charged species. Antibodies with more basic pI values are associated with increased tissue uptake and blood clearance, and vice versa for antibodies with more acidic pI values. Hence tissue distribution and pharmacokinetics (PK) can also be affected by charge heterogeneity.15 Most marketed therapeutic human or humanised antibodies have pI values of eight or above, which might be due to very similar frameworks.16,17
Serum stability
The highly complex nature of plasma can have a variety of effects on the physiochemical properties of antibodies, such as deamidation, methionine oxidation, cysteine pair shuffling, fragmentation, aggregation, and altered antigen binding.18 Many of these modifications can be appropriately assessed by characterisation of serum samples from in vivo experiments. For early-stage screening of candidates the in vivo conditions can be mimicked by incubating antibody in serum from relevant species (typically human, mouse and cynomolgus monkey) at 37C for a period of time. The samples can then be analysed for intact antigen binding properties by ELISA/flow cytometry or for specific amino acid modifications by mass spectrometry. The serum effects on self-association and aggregation can be measured in vitro by AC-SINS.19
Glycosylation
Glycosylation is another post-translational modification that can influence both the stability and the biological activity of an antibody and give rise to heterogeneity.20 Switching between expression host cell lines or changes in culturing conditions can alter the glycan profile.21 It is therefore important to ensure that the stability or activity of the antibody is not affected by this. Non-glycosylated antibody shows reduced Fc gamma receptor binding, lower thermostability, higher proteolytic susceptibility, propensity to aggregate, and structural changes compared to the glycosylated counterpart; hence the amount of non-glycosylated antibody should be kept to a minimum.20 On the other hand, glycans in the V-region can alter the antigen binding properties and so should generally be avoided. Glycan analysis (typically N- and O-linked glycosylation) can be carried out by capillary zone electrophoresis laser induced fluorescence detection (CZE-LIF) where the glycans are cleaved from the antibody, labelled with fluorophore, separated by CZE and identified by use of reference standards22 or by mass spectrometry (MS) based methods.23,24
Accelerated stress
Manufacturing, transport, storage and handling of therapeutics cause a great deal of physical and mechanical stress to the sample, which can affect the product’s stability; hence it is advantageous to identify candidates that are more resistant to stress. Procedures involving short-term accelerated stress to the sample have been developed to mimic these conditions. Commonly used stress factors are exposures to elevated temperatures, UV light, changes in pH, repeated freeze/thaw cycles, agitation and presence of oxidising agents.25 Testing the impact of some or all of these stress conditions on the biophysical stability of the antibody should be considered for all of the above parameters.
Criteria and limits
According to the International Council for Harmonisation (ICH) guidelines for biological products (Q6B and Q5C) and the European Medicines Agency (EMA) guideline on monoclonal antibodies, a combination of orthogonal methods must be used to thoroughly characterise the antibody’s biophysical profile. Individual or collective acceptance criteria and limits must be considered, proposed and justified for each evaluated property by the manufacturer (www.ich.org/home.html and www.ema.europa.eu/ema/).
The use of appropriate reference standards for each method can help to establish these criteria. An example of appropriate reference standards can be other well-characterised antibodies with known safety and stability profiles, such as marketed antibody drugs.26 The extensively characterised NISTmAb has recently been developed as a commercially available reference standard to provide a gold standard reference for biophysical characterisation in industry. In some cases it can also be helpful to have examples of antibodies with poor biophysical profiles to set the assay fail limits.
Surprisingly, despite extensive efforts to characterise protein aggregates, there are no regulatory limits for aggregates in biotherapeutics.2 The acceptance criteria for aggregate levels should reflect values that maintain the safety and efficacy of the product and must be set on a case-by-case basis. This is due to the uncertainty of how different aggregate species affect stability and safety.27 Although the guidelines have been written mainly for GMP purposes, considering the biophysical stability early in the lead discovery selection phase can help when selecting the best candidates and reduce the need for more extensive characterisation and/or engineering at later stages.
Finally, for a biologic the individual assays described in this article only give an indication of potential problems. The results from several of these assays must be considered collectively to form a stability-indicating profile. If the antibody fails to meet set criteria it must be considered if these liabilities can be improved by sequence engineering. Further information about cutting-edge technologies and developments can be found in this recent ACS book series: State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 1-3 (ISBN 9780841230262).
Biography
 DR LONE FRIIS qualified as a pharmacist from the Danish University of Pharmaceutical Sciences in Copenhagen, before obtaining a DPhil in clinical medicine from the University of Oxford at the MRC Weatherall Institute for Molecular Medicine. The research focussed on studying the structure-function relationship of T cells in multiple sclerosis. In 2010 she joined the BioTherapeutics group in MRC Technology’s Centre for Therapeutics Discovery (CTD), developing therapeutic antibodies. Lone is now head of the BioTherapeutics Protein Production and Characterisation Group delivering target proteins and antibodies for early-stage therapeutic antibody lead discovery.
DR LONE FRIIS qualified as a pharmacist from the Danish University of Pharmaceutical Sciences in Copenhagen, before obtaining a DPhil in clinical medicine from the University of Oxford at the MRC Weatherall Institute for Molecular Medicine. The research focussed on studying the structure-function relationship of T cells in multiple sclerosis. In 2010 she joined the BioTherapeutics group in MRC Technology’s Centre for Therapeutics Discovery (CTD), developing therapeutic antibodies. Lone is now head of the BioTherapeutics Protein Production and Characterisation Group delivering target proteins and antibodies for early-stage therapeutic antibody lead discovery.
 DR DAVID MATTHEWS completed his PhD in immunology at University College London, studying the functional relationship of IL-4 and IL-13, and subsequently did post-docs at UCL, Wurzburg University and the LMB in Cambridge UK, again studying the functional roles of IL-4 and IL-13. In 2001 he joined AERES Biomedical Ltd, a biotech specialising in antibody humanisation. In 2006 he joined the MRC Technology’s antibody engineering group, and in 2012 became the head of the BioTherapeutics Group. David and his team have been developing therapeutic antibodies to a wide range of targets in collaboration with academic and industrial partners.
DR DAVID MATTHEWS completed his PhD in immunology at University College London, studying the functional relationship of IL-4 and IL-13, and subsequently did post-docs at UCL, Wurzburg University and the LMB in Cambridge UK, again studying the functional roles of IL-4 and IL-13. In 2001 he joined AERES Biomedical Ltd, a biotech specialising in antibody humanisation. In 2006 he joined the MRC Technology’s antibody engineering group, and in 2012 became the head of the BioTherapeutics Group. David and his team have been developing therapeutic antibodies to a wide range of targets in collaboration with academic and industrial partners.
References
- Bee JS, Davis M, Freund E, Carpenter JF, Randolph TW. Aggregation of a monoclonal antibody induced by adsorption to stainless steel. Biotechnol Bioeng. 2010;105(1): 121-9
- Mahler HC, Friess W, Grauschopf U, Kiese S. Protein aggregation: pathways, induction factors and analysis. J Pharm Sci. 2009;98(9): 2909-34
- Niedziela-Majka A, Kan E, Weissburg P, Mehra U, Sellers S, Sakowicz R. High-throughput screening of formulations to optimize the thermal stability of a therapeutic monoclonal antibody. J Biomol Screen. 2015;20(4): 552-9
- Kelly RL, Sun T, Jain T, Caffry I, Yu Y, Cao Y, et al. High throughput cross-interaction measures for human IgG1 antibodies correlate with clearance rates in mice. MAbs. 2015;7(4): 770-7
- Hedberg SH, Heng JY, Williams DR, Liddell JM. Micro scale self-interaction chromatography of proteins: A mAb case-study. J Chromatogr A. 2016;1434: 57-63
- Liu Y, Caffry I, Wu J, Geng SB, Jain T, Sun T, et al. High-throughput screening for developability during early-stage antibody discovery using self-interaction nanoparticle spectroscopy. MAbs. 2014;6(2): 483-92
- Sun T, Reid F, Liu Y, Cao Y, Estep P, Nauman C, et al. High throughput detection of antibody self-interaction by bio-layer interferometry. MAbs. 2013;5(6): 838-41
- Jacobs SA, Wu SJ, Feng Y, Bethea D, O’Neil KT. Cross-interaction chromatography: a rapid method to identify highly soluble monoclonal antibody candidates. Pharm Res. 2010;27(1): 65-71
- Kohli N, Jain N, Geddie ML, Razlog M, Xu L, Lugovskoy AA. A novel screening method to assess developability of antibody-like molecules. MAbs. 2015;7(4): 752-8
- Jackisch C, Muller V, Maintz C, Hell S, Ataseven B. Subcutaneous Administration of Monoclonal Antibodies in Oncology. Geburtshilfe Frauenheilkd. 2014;74(4): 343-9
- Spencer S, Bethea D, Raju TS, Giles-Komar J, Feng Y. Solubility evaluation of murine hybridoma antibodies. MAbs. 2012;4(3): 319-25
- Yamniuk AP, Ditto N, Patel M, Dai J, Sejwal P, Stetsko P, et al. Application of a kosmotrope-based solubility assay to multiple protein therapeutic classes indicates broad use as a high-throughput screen for protein therapeutic aggregation propensity. J Pharm Sci. 2013;102(8): 2424-39
- Li L, Kantor A, Warne N. Application of a PEG precipitation method for solubility screening: a tool for developing high protein concentration formulations. Protein Sci. 2013;22(8): 1118-23
- Vlasak J, Ionescu R. Heterogeneity of monoclonal antibodies revealed by charge-sensitive methods. Curr Pharm Biotechnol. 2008;9(6): 468-81
- Boswell CA, Tesar DB, Mukhyala K, Theil FP, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug Chem. 2010;21(12): 2153-63
- Gahoual R, Beck A, Leize-Wagner E, Francois YN. Cutting-edge capillary electrophoresis characterization of monoclonal antibodies and related products. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1032: 61-78
- He XZ, Que AH, Mo JJ. Analysis of charge heterogeneities in mAbs using imaged CE. Electrophoresis. 2009;30(5): 714-22
- Correia IR. Stability of IgG isotypes in serum. MAbs. 2010;2(3): 221-32
- Li X, Geng SB, Chiu ML, Saro D, Tessier PM. High-throughput assay for measuring monoclonal antibody self-association and aggregation in serum. Bioconjug Chem. 2015;26(3): 520-8
- Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs. 2011;3(6): 568-76
- Miller AK, Hambly DM, Kerwin BA, Treuheit MJ, Gadgil HS. Characterization of site-specific glycation during process development of a human therapeutic monoclonal antibody. J Pharm Sci. 2011;100(7): 2543-50
- Zhang L, Lawson K, Yeung B, Wypych J. Capillary zone electrophoresis method for a highly glycosylated and sialylated recombinant protein: development, characterization and application for process development. Anal Chem. 2015;87(1): 470-6
- Reusch D, Haberger M, Falck D, Peter B, Maier B, Gassner J, et al. Comparison of methods for the analysis of therapeutic immunoglobulin G Fc-glycosylation profiles-Part 2: Mass spectrometric methods. MAbs. 2015;7(4): 732-42
- Song T, Ozcan S, Becker A, Lebrilla CB. In-depth method for the characterization of glycosylation in manufactured recombinant monoclonal antibody drugs. Anal Chem. 2014;86(12): 5661-6
- Alsaddique JA, Pabari RM, Ramtoola Z. Effect of Thermal and Shear Stressors on the Physical Properties, Structural Integrity and Biological Activity of the Anti-TNF-alpha Monoclonal Antibody, Infliximab. Curr Pharm Biotechnol. 2016;17(10): 905-14
- Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U S A. 2017;114(5): 944-9
- Vazquez-Rey M, Lang DA. Aggregates in monoclonal antibody manufacturing processes. Biotechnol Bioeng. 2011;108(7): 1494-508
Related topics
Antibodies, Drug Development, Protein, Therapeutics