Analysing recombinant proteins by mass spectrometry
Posted: 5 December 2015 | Claire Strain-Damerell (Structural Genomics Consortium), Nicola Burgess-Brown (Structural Genomics Consortium), Pravin Mahajan (Structural Genomics Consortium), Rod Chalk (Structural Genomics Consortium) | No comments yet
The drug discovery landscape is changing: no longer limited to big pharma, it is now within reach of academics and small consortia alike. Regardless of the setting, drug discovery requirements are always the same: strong biological theory, good chemical starting material and high quality protein samples from which to determine the binding and inhibition of the lead compounds…

Production of such high calibre protein is facilitated by a strong pipeline process (Figure 1), such as that established at the Structural Genomics Consortium (SGC), but ultimately there has to be a means to assess the quality of the protein before progressing to drug development. Traditionally this was limited to an approximate measure of mass and purity from SDS-PAGE and gel filtration profiles, however, advancements in mass spectrometry (MS) equipment and methodologies have greatly enhanced the breadth of information that is available from both pure and complex protein samples. MS can now be used to ask questions about protein folding, composition and modification state, to determine interactions between lead compounds or other proteins, and can even be used to predict the propensity for crystallisation. Here we discuss the application of MS at the SGC to aid production of proteins for structural and functional studies.
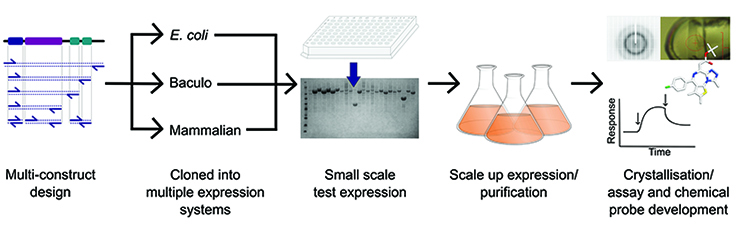
Figure 1: Overview of the protein production pipeline operated at the SGC.
The process of recombinant protein production involves quality checks throughout; on the expression constructs and also on their products. Verification of expression constructs by sequencing confirms identity at the DNA level but in addition to standard SDS-PAGE and gel filtration analyses, MS adds another dimension to protein analysis. Intact mass measurement, as well as confirming protein molecular weight, provides a quick indication of post-translational modifications (PTMs) such as phosphorylation, acetylation, glycosylation etc. Moreover, identification of proteins by tandem mass spectrometry (MS/MS) not only provides assurance on the protein of interest, but is also a valuable tool to identify host proteins that co-purify with recombinant proteins that could be mistaken as target proteins of interest.
Structural genomics laboratories apply MS as part of their protein production pipelines to provide information and guidance throughout the process1. Our process at the SGC begins at construct design; to increase the likelihood of obtaining soluble proteins and well-diffracting crystals, the multi-construct approach has been employed2,3. In order to implement this approach on multiple protein families, development of a high-throughput (HTP) protein expression and purification pipeline is required. This requirement has necessitated development of HTP expression methods utilising prokaryotic and eukaryotic systems3-6. Also, where possible, we apply HTP methods to our MS process to enable target protein identification for structural and functional studies as described below7.
Target protein identity
Use of SDS-PAGE alone as a measure of protein expression confirmation, monodispersity and identity based on mobility of protein bands is insufficient for several reasons: (i) poorly expressed proteins may not stand out clearly against the background of host proteins; (ii) target proteins may have anomalous gel mobility – for example membrane proteins and glycoproteins usually run faster than predicted and stain diffusely; (iii) the target protein may not actually have the expected sequence length – truncation or degradation are common resulting in faster gel mobility, while read-through to the next stop codon results in a larger than predicted protein bearing extraneous vector sequence; (iv) over-expression of host proteins such as heat shock proteins may appear due to the expression system itself and; (v) affinity capture can enrich for host proteins bearing sequence similarity to the affinity tag (Figure 2).
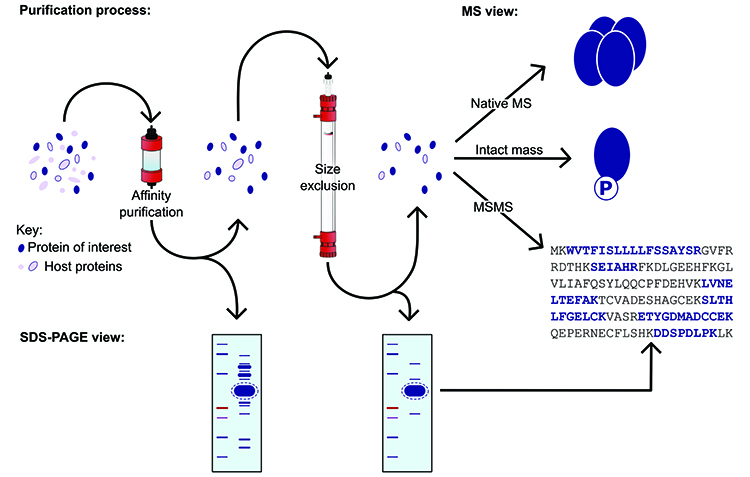
Figure 2: Illustration of the purification process, demonstrating the applications of mass spectrometry.
For all the above reasons, MS is rightly regarded as the ‘gold standard’ for protein identification, and this can be readily applied to HTP test expression8. Tryptic digest MS/MS analysis of SDS-PAGE gel bands is available in most laboratories and is sensitive to femtomoles of protein. Full sequence coverage is not required and confident identification of the target is possible from fragmentation data of a single peptide. Target bands can be precisely excised using gel-cutting tips, and the process of reduction-alkylation and tryptic digestion can be performed on a large number of samples in parallel using 96-well plates and multichannel pipettes. Fast LC-MS/MS analysis protocols and automated database searching means that MS/MS identification of all gel bands from a 96-well test expression is possible in under 72 hours7. In a HTP environment and in the absence of MS, the risk of misidentification of expressed soluble proteins is in the region of 10%, whereas for membrane proteins the risk is as high as 50%. Incorporation of tryptic digest MS/MS into a HTP pipeline enables selection of constructs for scale-up with confidence that the target is indeed present.
Target protein structure
While tryptic digest MS/MS can confirm target identity, it cannot confirm covalent structure because, for technical reasons, 100% peptide coverage is never achieved. Truncated and full length constructs may generate fragment spectra from the same subset of tryptic peptides and therefore yield identical Mascot results. PTMs such as terminal methionine loss and acetylation may be naturally present, and others may have been artificially introduced, such as biotinylation. These PTMs will not come to light if they are absent from the database being searched, which is normally the case. In general, results for tryptic digest MS/MS of cut gel bands are not available in less than 24 hours. Techniques exist for rapid trypsinisation9 and top-down fragmentation10 but these are not immediately available to the scientist performing protein purification at the bench. It is well known that speed and success go together for purification of native proteins. Having purified a protein, the preparation cannot wait days for the results of MS analysis to become available.
 Protein intact mass analysis is an extremely fast method for determining covalent structure and is also simple enough to be performed by bench scientists with minimal MS training on an open access basis. Typically a microgram of purified protein is diluted in an acidic buffer and undergoes LC-MS analysis using a guard column as a protein trap and a reversed-phase elution over less than two minutes7. Data analysis involves summing together of mass to charge (m/z) spectra over the retention time of the relevant peak and mathematical transformation of the multiple charge states of the electrospray spectrum to a single neutral peak representing the observed protein mass. This can be fully automated, leaving the experimenter to compare the observed mass with that expected from the construct sequence and to interpret any mass deviations. In principle, MALDI MS can be used in this way, but in practice the necessary sensitivity and mass accuracy of less than 1 Da is only achievable using electrospray. Where observed and expected masses are within 1 Da, this is sufficient to confirm both the identity and structure of the target protein. A decreasing series of sodium adduct peaks is characteristically observed which has no functional effect upon the protein.
Protein intact mass analysis is an extremely fast method for determining covalent structure and is also simple enough to be performed by bench scientists with minimal MS training on an open access basis. Typically a microgram of purified protein is diluted in an acidic buffer and undergoes LC-MS analysis using a guard column as a protein trap and a reversed-phase elution over less than two minutes7. Data analysis involves summing together of mass to charge (m/z) spectra over the retention time of the relevant peak and mathematical transformation of the multiple charge states of the electrospray spectrum to a single neutral peak representing the observed protein mass. This can be fully automated, leaving the experimenter to compare the observed mass with that expected from the construct sequence and to interpret any mass deviations. In principle, MALDI MS can be used in this way, but in practice the necessary sensitivity and mass accuracy of less than 1 Da is only achievable using electrospray. Where observed and expected masses are within 1 Da, this is sufficient to confirm both the identity and structure of the target protein. A decreasing series of sodium adduct peaks is characteristically observed which has no functional effect upon the protein.
Large negative mass shifts can often be accounted for by truncation of the target protein. These will match a C-terminal or N-terminal sequence string to within 1 Da and will be inclusive of any MS/MS peptide data. Most PTMs involve mass additions. Serial additions, as observed in phosphorylation and glycosylation, are simple to interpret. While there are just under a thousand PTMs in the Unimod database (http://www.unimod.org)11, the vast majority are extremely rare, such as mutation, or involve chemical derivitisation or heavy isotopes. Excepting glycans, there are actually fewer than a dozen modifications routinely observed in E. coli, insect and mammalian expression systems. Parallel mass additions involving more than one modification can be more difficult to interpret, but knowledge of PTMs which commonly occur together, such as methionine loss and acetylation still makes this possible.
Like soluble proteins, integral membrane proteins are also amenable to intact mass analysis, though different methods are required. The main difficulty involves separation of the protein from detergent, which is a powerful ion suppressant. This may be done either off-line using size exclusion or protein precipitation, or on-line using reversed phase column separation12. Techniques also exist for gas-phase separation of the protein-detergent complex in the mass spectrometer itself by collision-induced dissociation13.
The same high-throughput methods which can be applied to tryptic digest MS/MS may also be applied to intact mass analysis at the small-scale test expression stage. Proteins from 96-well test purification may be analysed overnight with results available the following day. Unlike the former technique, intact mass analysis is insufficiently sensitive for targets expressing with low to medium yield, yet this need not be of concern since often highly expressing constructs are preferred for scale-up. When intact mass data can be obtained, this is available faster and is more informative during the protein purification process than tryptic digest MS/MS.
Target protein function
Functionally-active protein is the goal of most recombinant protein expression. Functional activity of an enzyme can be measured indirectly by quantitative mass spectrometric analysis of the conversion of substrate to product14. Where biological activity involves specific binding of a small molecule ligand, interaction with another protein, or formation of a multimeric protein complex, this can be measured directly using native MS15. Even when the activity of a protein is unknown, the proportion of correctly folded native protein may be determined and hence its functional activity inferred. The methods discussed earlier involve loss of all activity either by proteolysis or by denaturing HPLC. Native MS involves achieving ionisation whilst allowing the protein to retain its functional conformation.
Efficient desalting is performed off-line and proteins are introduced into the mass spectrometer by direct infusion. Natively folded proteins acquire two-to-three-fold fewer charges upon ionisation, hence the m/z ratio (the parameter which mass spectrometry actually measures) will be higher. For small proteins m/z will fall within the normal range of most instruments of up to m/z 3500. Larger proteins and protein complexes with a higher native m/z will require a modern mass spectrometer which can operate to m/z 20,000 and above. The transmission and detection efficiency falls away as m/z increases, meaning that some optimisation of the instrument is needed for native MS. In spite of this, the resolution and mass accuracy obtainable by native MS can equal or even surpass that seen using conventional methods.
At the SGC, all of these analyses are used together to build a complete picture of what our protein samples consist of and how they behave. This allows independent purifications to be compared and normalised for consistency, giving weight and reliability to the downstream uses in structural biology, as well as assay and chemical probe development.
Acknowledgements: This work was supported by the SGC which is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust [092809/Z/10/Z].
Biography
 NICOLA BURGESS-BROWN is the Principal Investigator of the Biotech Group at the SGC, responsible for molecular biology, cell culture, protein production and mass spectrometry analysis of the targets of interest at the Oxford site. Working closely with other SGC teams, the group develops methods for increasing protein expression and driving throughput. Following her degree in Applied Biochemical Sciences in 1997, Nicola worked as a Molecular Biologist for SmithKline Beecham. She received her PhD in Molecular Microbiology at the University of Nottingham in 2001 then returned to industry focusing on high-throughput cloning and validation of therapeutic cancer antigens for Oxford Glycosciences.
NICOLA BURGESS-BROWN is the Principal Investigator of the Biotech Group at the SGC, responsible for molecular biology, cell culture, protein production and mass spectrometry analysis of the targets of interest at the Oxford site. Working closely with other SGC teams, the group develops methods for increasing protein expression and driving throughput. Following her degree in Applied Biochemical Sciences in 1997, Nicola worked as a Molecular Biologist for SmithKline Beecham. She received her PhD in Molecular Microbiology at the University of Nottingham in 2001 then returned to industry focusing on high-throughput cloning and validation of therapeutic cancer antigens for Oxford Glycosciences.
 ROD CHALK is a post doc running the mass spectrometry facility at the SGC, where he has worked since 2008. Rod gained a PhD at the Liverpool School of Tropical Medicine in 1992 and has worked in mass spectrometry in industry and academia for 19 years in a variety of roles. Industrial positions include Oxford Glycosciences (proteomics), and Comet Analytics (cryodetector mass spectrometry) and Lonza (biologics). He held academic posts in proteomics at QUB, Oxford and Reading. His current interests include high-throughput protein analysis, integral membrane proteins and native mass spectrometry and their application to drug discovery.
ROD CHALK is a post doc running the mass spectrometry facility at the SGC, where he has worked since 2008. Rod gained a PhD at the Liverpool School of Tropical Medicine in 1992 and has worked in mass spectrometry in industry and academia for 19 years in a variety of roles. Industrial positions include Oxford Glycosciences (proteomics), and Comet Analytics (cryodetector mass spectrometry) and Lonza (biologics). He held academic posts in proteomics at QUB, Oxford and Reading. His current interests include high-throughput protein analysis, integral membrane proteins and native mass spectrometry and their application to drug discovery.
 After studying Molecular Genetics in Biotechnology at the University of Sussex DR CLAIRE STRAIN-DAMERELL continued on to obtain her DPhil on the role of the redox sensitive transcriptional repressor; Rex, in Streptomyces coelicolor. After completing her PDhil Dr Strain-Damerell then progressed on to a postdoctoral position at the Structural Genomics Consortium in Oxford, focusing on the optimisation of the cloning and crystallographic pipelines to improve the success rates for structural determination.
After studying Molecular Genetics in Biotechnology at the University of Sussex DR CLAIRE STRAIN-DAMERELL continued on to obtain her DPhil on the role of the redox sensitive transcriptional repressor; Rex, in Streptomyces coelicolor. After completing her PDhil Dr Strain-Damerell then progressed on to a postdoctoral position at the Structural Genomics Consortium in Oxford, focusing on the optimisation of the cloning and crystallographic pipelines to improve the success rates for structural determination.
 PRAVIN MAHAJAN obtained his PhD from De Montfort University, Leicester, where he studied genetic engineering of drug-metabolising human Cytochrome P450 enzymes and their interaction with NADPH-cytochrome P450 reductase. Prior to his PhD, he worked in the area of cancer drug discovery for five years. Pravin joined the SGC in 2008 and has been working on high-throughput expression of human proteins in mammalian cells, insect cells and E. coli. He developed a high-throughput method for protein expression in mammalian cells using BacMam and contributed significantly towards method development and improvement of the baculovirus expression system.
PRAVIN MAHAJAN obtained his PhD from De Montfort University, Leicester, where he studied genetic engineering of drug-metabolising human Cytochrome P450 enzymes and their interaction with NADPH-cytochrome P450 reductase. Prior to his PhD, he worked in the area of cancer drug discovery for five years. Pravin joined the SGC in 2008 and has been working on high-throughput expression of human proteins in mammalian cells, insect cells and E. coli. He developed a high-throughput method for protein expression in mammalian cells using BacMam and contributed significantly towards method development and improvement of the baculovirus expression system.
References
- Jeon WB, Aceti DJ, Bingman CA, Vojtik FC, Olson AC, Ellefson JM, et al. High-throughput purification and quality assurance of Arabidopsis thaliana proteins for eukaryotic structural genomics. Journal of structural and functional genomics. 2005;6(2-3):143-7
- Graslund S, Sagemark J, Berglund H, Dahlgren LG, Flores A, Hammarstrom M, et al. The use of systematic N- and C-terminal deletions to promote production and structural studies of recombinant proteins. Protein Expr Purif. 2008;58(2):210-21
- Savitsky P, Bray J, Cooper CD, Marsden BD, Mahajan P, Burgess-Brown NA, et al. High-throughput production of human proteins for crystallization: the SGC experience. J Struct Biol. 2010;172(1):3-13
- Strain-Damerell C, Mahajan P, Gileadi O, Burgess-Brown NA. Medium-throughput production of recombinant human proteins: ligation-independent cloning. Methods Mol Biol. 2014;1091:55-72
- Burgess-Brown NA, Mahajan P, Strain-Damerell C, Gileadi O, Graslund S. Medium-throughput production of recombinant human proteins: protein production in E. coli. Methods Mol Biol. 2014;1091:73-94
- Mahajan P, Strain-Damerell C, Gileadi O, Burgess-Brown NA. Medium-throughput production of recombinant human proteins: protein production in insect cells. Methods Mol Biol. 2014;1091:95-121
- Chalk R, Berridge G, Shrestha L, Strain-Damerell C, Mahajan P, Yue W, et al. High-Throughput Mass Spectrometry Applied to Structural Genomics. Chromatography. 2014;1(4):159-75
- Cohen SL, Chait BT. Mass spectrometry as a tool for protein crystallography. Annual review of biophysics and biomolecular structure. 2001;30(1):67-85
- Sebela M, Stosova T, Havlis J, Wielsch N, Thomas H, Zdrahal Z, et al. Thermostable trypsin conjugates for high-throughput proteomics: synthesis and performance evaluation. Proteomics. 2006;6(10):2959-63
- Brunner AM, Lossl P, Liu F, Huguet R, Mullen C, Yamashita M, et al. Benchmarking multiple fragmentation methods on an orbitrap fusion for top-down phospho-proteoform characterization. Analytical chemistry. 2015;87(8):4152-8
- Creasy DM, Cottrell, JS. Unimod: Protein modifications for mass spectrometry. Proteomics. 2004;4:1534–6
- Berridge G, Chalk R, D’Avanzo N, Dong L, Doyle D, Kim JI, et al. High-performance liquid chromatography separation and intact mass analysis of detergent-solubilized integral membrane proteins. Analytical biochemistry. 2011;410(2):272-80
- Barrera NP, Isaacson SC, Zhou M, Bavro VN, Welch A, Schaedler TA, et al. Mass spectrometry of membrane transporters reveals subunit stoichiometry and interactions. Nature methods. 2009;6(8):585-7
- Forbes CD, Toth JG, Ozbal CC, Lamarr WA, Pendleton JA, Rocks S, et al. High-throughput mass spectrometry screening for inhibitors of phosphatidylserine decarboxylase. Journal of biomolecular screening. 2007;12(5):628-34
- Heck AJ. Native mass spectrometry: a bridge between interactomics and structural biology. Nature methods. 2008;5(11):927-33
Related topics
Analytical Techniques, Drug Discovery, Genomics, Mass Spectrometry, Protein, Protein Expression