If Columbus had a map… guiding the discovery of new drugs
Posted: 4 June 2020 | Horst Flotow (GDBspace), Jingwen Shi (GDBspace), Sacha Javor (GDBspace) | No comments yet
Exploring large databases and selecting compounds of interest can be extremely time-consuming for researchers. Here, Sacha Javor, Horst Flotow and Jingwen Shi discuss a novel chemoinformatics approach for drug discovery.

Geographic maps have helped us to navigate the Earth’s surface since antiquity, while thematic maps inform us about the world by providing more granularity on features connected with a specific geographic area, such as rainfall distribution or population density. What if there was such a map for chemical space and its features, particularly ones that apply to drug discovery?
In chemistry, there is already a map describing all the known chemical elements – the periodic table. Mendeleev’s organisation of the periodic table was used to derive relationships between the various elements, predict chemical properties and even the existence of yet undiscovered elements. As elements of the table make up all known molecules, how can we drill down from the periodic table to a map of molecules (and the chemical space they occupy) that can help medicinal chemists navigate and predict the relationships and properties of molecules instead of simple elements? As a matter of fact, chemical “cartographers” in Switzerland have recently created such a map.
Drug discovery is frequently likened to finding a needle in a haystack, where the screening of millions of candidates often leads to just one drug. Many researchers resort to virtual screening or artificial intelligence (AI)-driven predictive methods to boost their drug discovery efforts, conducting part of the needle-finding exercise using a computer. Professor Jean-Louis Reymond and team (computer scientist Dr Daniel Probst and computational chemist Dr Sacha Javor, among others) at the University of Bern in Switzerland have taken an interesting perspective to tackle drug discovery – namely using a map to guide the discovery of new drugs1-4 (Figure 1). This technology has since been transferred to GDBspace Ltd, where it is being refined and made available for drug discovery.
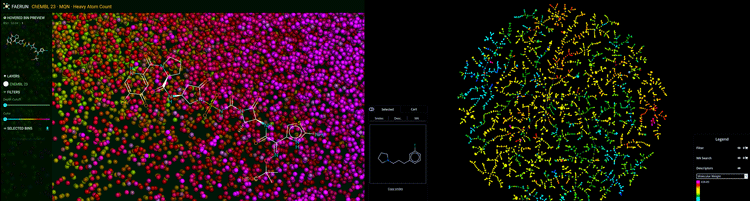
Figure 1: Chemical space of molecules visualised in 3D (left) and in 2D as a tree map (right). Predictive visualisation by GDBspace Ltd.
How does this work?
Molecules are first retrieved from very large inhouse molecular databases.5-8 The software then computes a map of molecules based on their “molecular fingerprint”. The unique fingerprints, some of which were specifically developed for that purpose, provide a high-resolution indexing of the molecules according to their substructures,9 allowing near-neighbour searches and/or similarity searches of molecules.4 This technique provides an intuitive representation of the orientation and location in chemical space for all these molecules. In addition, the method can be extended to a multi‑fingerprint approach and applied when predicting potential on- and off-target effects, by examining maps and determining if a newly identified bioactive molecule is closely related to (ie, is a near neighbour of) molecules with documented bioactivity or toxicity. The proximity to such known bioactives indicates likely interaction with the corresponding biological targets.10-12 As Professor Reymond says: “This technology works very broadly for small molecule and peptide drug discovery. The map can be configured to include sophisticated virtual screening or machine learning algorithms. This approach allows users to understand and literally see the result of these advanced and often cutting-edge predictive methods. Instead of receiving a long list of results, followed by an iterative cycle of designing, synthesising and experimentally testing molecules for activity, one may be able to pick molecules that are near neighbours of a target and only test those in one cycle. This can potentially save significant resources and time and prevent researchers from getting stuck in the wrong corner of chemical space (the vast universe of small organic molecules that might exhibit drug activity is estimated to be in the order of 1060 molecules).”
Consider an analogy…
If you receive a list of 200 countries, it would be a challenge for anyone’s brain to remember, make sense of and connect the dots. However, equipped with a world map, not only can you remember numerous countries, you can also uncover additional insights: Which countries are on the same continent, climate zone and situated next to each other? Which countries have similarities such as size, shape or length of their coastline? What new insights and patterns can be discovered if you overlay the world map with the per capita GDP (Gross Domestic Product)? – just to name a few examples. Another advantage of the map is that you can search (requiring predefined knowledge) and browse (requiring no predefined knowledge), ie, discover new information about countries and organise the information in new ways to gain valuable insights.
Why is that? According to human brain research, the brain processes visual information 60,000 times faster than text and 90 percent of information transmitted to the brain is visual while roughly 65 percent of the populations are visual learners. In short, humans are visual creatures. While “big data” and “AI” are becoming buzzwords, we tend to forget that as datasets further increase in size and complexity, individuals, as well as organisations, are increasingly drowning in data. This is where advanced visualisation comes into play, combining the best of three worlds – experimental data, computer power and the human brain. It is not trivial to leverage the power of the human brain, enabling human intuition at superhuman scale as well as empowering better predictions and decision-making – an area often underestimated in the era of AI.
Case study – COVID-19
In the race to discover COVID-19 inhibitors, Ruibeixiyu Biotech leveraged GDBspace’s computational technologies and knowhow to preselect molecules of interest. Experimental validations of six suggested, synthesised and tested molecules provided one with nanomolar activity (comparable in vitro activity as the antiviral medication Remdesivir developed by the American biopharmaceutical company Gilead Sciences). Thus, in the race to develop novel antivirals to combat coronavirus infections and the spread of the virus, this technology cut the project time down to a matter of days, when this usually takes months. By increasing the speed and lowering the cost of drug discovery, utilising software has the potential to meet the acute needs of patients in the latest coronavirus epidemic outbreak, as well as other major unmet medical challenges, in the timeliest fashion.
Where will this technology take drug discovery?
In addition to the case study described above, an increasing number of pharmaceutical companies are using this technology to accelerate their drug discovery efforts and shorten the time taken to develop novel medicines. The cheminformatic market potential is estimated to grow at a compound annual growth rate of 18.7 percent, to $21.2 billion by 2024.

Although the current focus is on the pharmaceutical industry and drug discovery, there are a number of additional potential applications of this fingerprinting and mapping technology that reaches beyond cheminformatics. One such area may be in automated image analysis for high-content screening. Here, this unbiased fingerprinting and analysis may be coupled to other AI and machine-learning techniques and be useful in understanding the hundreds of features that can already be extracted from the images but not yet exploited to their full extent. Organising the data into maps might help to unravel previously unknown relationships and derived features. Such an approach, which makes use of vastly increased amounts of information, is potentially more reliable than the current approaches that often rely on expert knowledge.
It is of course always possible to make the greatest breakthrough discoveries without a map, but arguably, if Colombus had a map, the world would be a very different place today.
About the authors

Jingwen Shi has a PhD from Karolinska Institute, Sweden and is a visiting scholar at Max-Planck Institute for Plantphysiology in Germany and Pittsburgh University in USA. She has published her research in several top journals and is the co-founder of GDBspace Ltd.

Horst Flotow is a biochemist with many years of experience working in the pharmaceutical industry. In Singapore, he was a founding Group Leader and Head of the Singapore Screening Centre at the A-STAR’s Experimental Therapeutics Centre. More recently, he has lead Hit Discovery Constance in Germany and is now a Senior Director of Business Development and Research Collaboration at HitGen Inc.

Sacha Javor is a computational and organic chemist with over 15 years’ experience in chemoinformatics applied to drug discovery. He is the author of numerous studies in major international peer reviewed journals. Sacha is an EPFL alumni and has a PhD from the University of Bern, Switzerland. He is also an expert in translational and entrepreneurial medicine with a MAS from the SITEM-Insel, Bern. He has spent several years in biomedical research at The Scripps Research Institute in La Jolla, CA.
References
1. Awale M, Reymond JL. Exploring chemical space for drug discovery using the chemical universe database, ACS Chem. Neurosci., 2012, 3, 649-657.
2. Probst D, Reymond JL. Visualization of very large high-dimensional datasets as minimum spanning trees, J. Cheminformatics, 2020, doi:10.1186/s13321-020-0416-x
3. Probst D, Reymond JL, FUn: a framework for interactive visualizations of large, high-dimensional datasets on the web, Bioinformatics, 2017, 34 (8), 1433-1435.
4. Delalande C, Awale M, Rubin M, Probst D, Ozhathil LC, Gertsch J, Abriel H, Reymond JL. Optimizing TRPM4 inhibitors in the MHFP6 chemical space. Eur. J. Med. Chem., 2019, 166, 167-177.
5. Awale M, Visini R, Probst D, Arús-Pous J, Reymond JL. Chemical space: big data challenge for molecular diversity. JL. Chimia, 2017, 71 (10), 661-666.
6. Awale M, van Deursen R, Reymond JL. MQN-mapplet: visualization of chemical space with interactive maps of DrugBank, ChEMBL, PubChem, GDB-11, and GDB-13. J. Chem. Inf. Model. 2013, 53, 509-518.
7. Bühlmann S, Reymond JL. ChEMBL-Likeness Score and Database GDBChEMBL. Front. Chem, 2020, doi:10.3389/fchem.2020.00046
8. Meier K, Bühlmann S, Arús-Pous J, Reymond JL. Chimia, 2020, doi:10.2533/ chimia.2020.241
9. Probst D, Reymond JL. A probabilistic molecular fingerprint for big data settings. J. Cheminformatics. 2018. 66 (10), doi:10.1186/s13321-018-0321-8
10. Awale M, Reymond JL. Polypharmacology Browser PPB2: Target prediction combining nearest neighbors with Machine Learning. J. Chem. Inf. Model., 2018, doi:10.1021/acs.jcim.8b00524
11. Aware M, Reymond JL. The polypharmacology browser: a web-based multi-fingerprint target prediction tool using ChEMBL bioactivity data. J. Cheminform., 2017, 9, 11.
12. Poirier M, Awale M, Roelli M, Giuffredi G, Ruddigkeit L, Evensen L, Stooss A, Calarco S, Lorens J, Charles RP, Reymond JL. ChemMedChem, 2019, 14 (2), 224-236.
Related topics
Analysis, Analytical Techniques, Artificial Intelligence, Big Data, Drug Discovery, Drug Leads, Molecular Biology, Molecular Targets, Technology
Related conditions
Coronavirus, Covid-19
Related organisations
ForteBio, GBDspace, Gilead Sciences Inc, Horizon Discovery, Merck, Ncardia, Ruibeixiyu Biotech, University of Bern
Related people
Professor Jean-Louis Reymond