RNA folding: new model could change future drug design
Posted: 5 November 2025 | Drug Target Review | No comments yet
A Japanese research team has simulated how RNA molecules fold, using cutting-edge computational tools to model complex structures with accuracy – a breakthrough that could accelerate the development of RNA-based medicines and therapies.

A Japanese research team has achieved a new milestone in understanding how ribonucleic acid (RNA) molecules fold – a process central to the development of RNA-based therapeutics.
The study, led by associate professor Tadashi Ando from the Department of Applied Electronics at Tokyo University of Science has demonstrated that advanced computational methods can accurately model the folding of a wide range of RNA structures.
Unlocking RNA’s structural secrets
RNA is a very versatile molecule, involved in regulating, processing and maintaining genetic information across all living systems. Its ability to adopt complex three-dimensional shapes – known as secondary and tertiary structures – is crucial to its function.
Automation now plays a central role in discovery. From self-driving laboratories to real-time bioprocessing
This report explores how data-driven systems improve reproducibility, speed decisions and make scale achievable across research and development.
Inside the report:
- Advance discovery through miniaturised, high-throughput and animal-free systems
- Integrate AI, robotics and analytics to speed decision-making
- Streamline cell therapy and bioprocess QC for scale and compliance
- And more!
This report unlocks perspectives that show how automation is changing the scale and quality of discovery. The result is faster insight, stronger data and better science – access your free copy today
RNA is a very versatile molecule, involved in regulating, processing and maintaining genetic information across all living systems.
With the fast rise of RNA-based therapeutics, from vaccines to gene-silencing drugs, researchers are increasingly focused on understanding and predicting how RNA folds. Yet simulating the complete folding process of an RNA molecule from an unfolded state remains one of computational biophysics’ greatest challenges.
Traditional molecular dynamics (MD) simulations use intricate physics-based models, known as force fields, to predict how molecules behave. However, accurate folding simulations have largely been limited to a handful of small, simple RNA stem-loop structures.
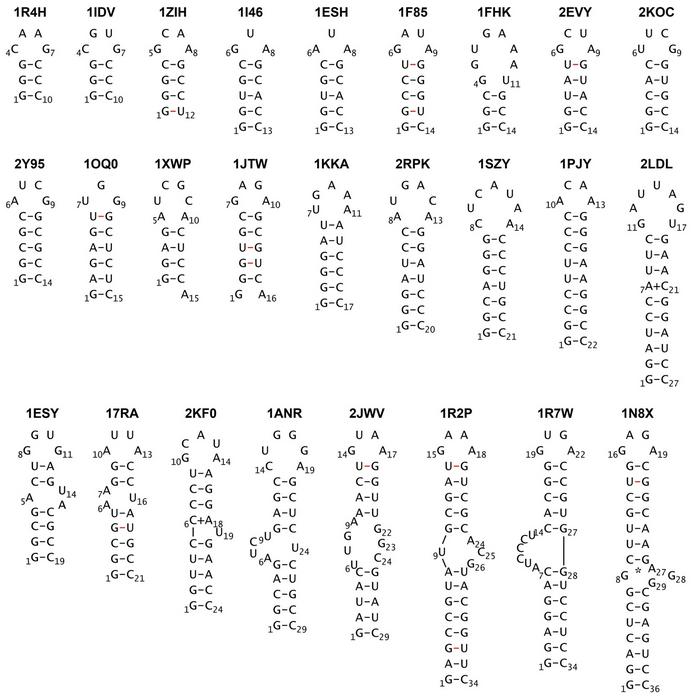
This image depicts the base pairs and simplified structure of the stem loops simulated in this study. Credit: Associate Professor Tadashi Ando from Tokyo University of Science, Japan.
Testing cutting-edge computational tools
Against this backdrop, Dr Ando conducted a large-scale and rigorous assessment of modern simulation techniques to determine whether today’s computational tools could handle a far more diverse and complex range of RNA structures.
Dr Ando conducted a large-scale and rigorous assessment of modern simulation techniques to determine whether today’s computational tools could handle a far more diverse and complex range of RNA structures.
His research integrated two cutting-edge components. The first was the DESRES-RNA atomistic force field – a finely tuned potential function designed for accurate RNA simulations. The second was the GB-neck2 implicit solvent model, which greatly accelerates computations by representing the surrounding liquid as a continuous medium, rather than modelling each water molecule individually.
Dr Ando applied this combination to 26 RNA stem-loops, ranging from 10 to 36 nucleotides in length, including structures with bulges and internal loops. Each simulation began from a fully extended, unfolded conformation.
Folding accuracy surpasses expectations
The combined DESRES-RNA and GB-neck2 methods achieved accurate folding for 23 of the 26 RNA molecules tested.
For the 18 simpler stem-loops, the simulations reproduced their expected structures with strong accuracy, achieving root mean square deviation (RMSD) values of less than 2 Å for the stems and less than 5 Å overall. Even among more challenging structures containing bulges or internal loops, five of the eight were successfully folded.
“Previous studies have reported only two or three folding examples of simple stem-loop structures of approximately 10 residues, whereas this study deals with structures of much greater scale and complexity,” said Dr Ando.
Insights and future improvements
While the simulations demonstrated strong predictive power, Dr Ando noted that further refinements are needed. The loop regions of the RNA molecules showed slightly less accuracy, with RMSD values around 4 Å, suggesting that the implicit solvent model requires tuning to better account for non-canonical base pairs and the effects of divalent cations such as magnesium.
The study nevertheless represents a significant milestone in RNA modelling. It validates the use of modern force fields and solvent models as reliable tools for exploring RNA conformational changes on a large scale.

These images draw a comparison between the simulated folding and experimentally determined structures of RNA stem loops. Credit: Associate Professor Tadashi Ando from Tokyo University of Science, Japan.
Implications for medicine and drug design
Understanding RNA folding at this level has far-reaching implications for biotechnology and medicine. By accurately modelling how RNA molecules fold, researchers can design more effective RNA-based drugs – potentially leading to new treatments for genetic disorders, viral infections such as COVID-19, influenza and certain cancers.
“The ability to reproduce the overall folding of the basic stem-loop structure is an important milestone in understanding and predicting the structure, dynamics and function of RNA using highly accurate and reliable computational models,” said Dr Ando. “I expect this computational technique to lead to applications in RNA molecule design and drug discovery.”
Related topics
Analysis, Computational techniques, Drug Discovery, Drug Discovery Processes, Molecular Modelling, Structural Biology, Translational Science
Related conditions
Cancer, Covid-19, genetic disorders, Influenza, viral infections
Related organisations
the Department of Applied Electronics at Tokyo University of Science