How ageing triggers Huntington’s disease
Posted: 7 November 2022 | Ria Kakkad (Drug Target Review) | No comments yet
A new study found that as patients age, Huntington’s disease gradually impairs the important cellular housekeeping process autophagy, which is responsible for eliminating waste from cells.
Huntington’s disease, a fatal, inherited neurodegenerative condition, is caused by a genetic error present at birth, though its symptoms often do not begin until middle adulthood. Scientists at Washington University School of Medicine in St Louis, US, have been trying to understand how the ageing process triggers the onset of symptoms, with the expectation that such knowledge could point to treatments that delay or prevent neurodegeneration.
To that end, a new study from Washington University indicates that as patients age, the disease gradually impairs an important cellular housekeeping process called autophagy, which is responsible for eliminating waste from cells. This housekeeping is significant in Huntington’s because a build-up of waste in a specific kind of neuron leads to such cells’ untimely deaths.
The researchers also showed that enhancing the autophagy pathway in such neurons that were created from skin cells of Huntington’s patients protects those cells from dying.
“Our study reveals how ageing triggers a loss of the crucial process of autophagy — and hints at how we might try to restore this important function, with the aim of delaying or even preventing Huntington’s disease,” said senior author Dr Andrew Yoo.
The study, which was published in Nature Neuroscience, also may offer clues to understanding cognitive decline in ageing generally.
Huntington’s disease destroys a specific type of brain cell called medium spiny neurons, the loss of which causes involuntary muscle movements, impaired mental health and cognitive decline. Patients typically live about 20 years after signs of the disease first appear.
We wanted to find the difference between young patients who have no symptoms and older patients who actively show signs of the disease”
For this study, the researchers reprogrammed patients’ skin cells into medium spiny neurons using a technique they developed that allows adult skin cells to be transformed directly into various types of brain cells, depending on the specific recipe of signalling molecules to which the skin cells are exposed. More common techniques involve use of stem cells — but stem cells reset the cells’ biological clocks to an early developmental state, which is not useful when studying diseases that only become symptomatic in adulthood.
“We collected skin cell samples from different patients at a range of ages and modelled the disease before and after symptoms developed, which allowed us to identify the differences between younger and older patients with Huntington’s disease,” Yoo said. “We knew there must be some change that takes place as patients age. They all have a genetic mutation in the Huntington gene. We wanted to find the difference between young patients who have no symptoms and older patients who actively show signs of the disease.”
The team found that medium spiny neurons reprogrammed from skin cells of older patients with symptomatic Huntington’s produced very high levels of a microRNA molecule called miR-29b-3p. These high levels were not seen in reprogrammed neurons of younger Huntington’s patients or in reprogrammed neurons from healthy individuals of any age. The investigators showed that the microRNA set off a chain of events that included impairing autophagy in these cells. When the skin cells completed the conversion into neurons, they began producing the problematic microRNA, autophagy slowed down and the cells began dying.
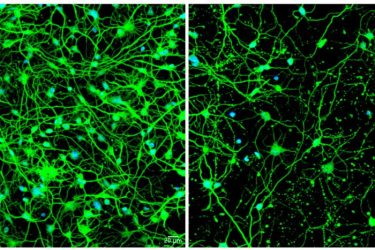
A study from Washington University School of Medicine in St Louis shows that as patients age, Huntington’s disease impairs autophagy, which eliminates waste from cells. Shown at left are neurons transformed from skin cells of a young patient with pre-symptomatic Huntington’s disease. On the right are neurons transformed from skin cells of an older patient with symptomatic Huntington’s; these cells are sparse because the ageing process impairs autophagy, leading to cell death
[Credit: Youngmi Oh, Seongwon Lee].
The researchers went on to show that reducing levels of this microRNA allowed autophagy to continue and protected the neurons from dying. In addition, they found that enhancing autophagy with a chemical compound called G2 protected the diseased neurons from death. As the researchers increased the dose of G2, the protection from cell death improved as well.
G2 is derived from a series of analogy that were discovered. G2 was identified via high-throughput screening for autophagy enhancer drugs that could correct the cellular accumulation of variant alpha-1-antitrypsin Z that causes liver disease in alph-1-antitrypsin deficiency (ATD). The G2 compounds could therefore represent attractive candidates for preventing neurodegeneration in Huntington’s disease, liver disease in alpha-1-antitrypsin deficiency and perhaps other diseases in which aberrant accumulation of misfolded proteins is toxic to cells.
The study also uncovered what may be a tantalising clue for understanding cognitive decline in normal ageing. When comparing the symptomatic neurons to pre-symptomatic neurons and to healthy neurons from both young and older adults, the researchers found that the neurons of healthy older adults produced slightly elevated levels of the harmful microRNA, but in far smaller amounts than the neurons of symptomatic Huntington’s disease patients. The study suggests that even in normal, healthy ageing, medium spiny neurons gradually produce low levels of this microRNA, which may interfere with autophagy’s healthy cellular housekeeping.
“By modelling different stages of the disease across the life span, we can identify how aging plays a role in disease onset,” Yoo said. “With that information, we can begin to look for ways to delay that onset. Our study also suggests that the triggering molecule for the onset of Huntington’s disease may play some role in age-associated decline in neuronal function generally. Understanding the component of aging that sets off neurodegeneration may help in developing new strategies for treatment and prevention of Huntington’s disease and other neurodegenerative conditions that develop at older ages.”
The team also are working with other collaborators using their cellular reprogramming technique to investigate forms of Alzheimer’s disease, tauopathy and other neurodegenerative conditions.
Related topics
Disease Research, Neurons, Neurosciences, RNAs
Related conditions
Huntington's disease
Related organisations
Washington University School of Medicine
Related people
Dr Andrew S. Yoo