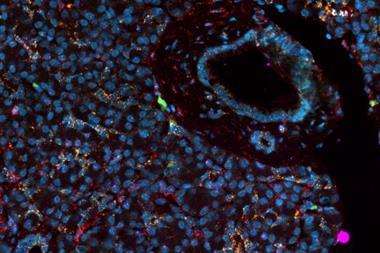
In the sections that follow, we dive in on the latest research describing the underlying causes of ALS and a closely related condition known as frontotemporal dementia (FTD), two forms of neurodegenerative disease that share striking similarities in their molecular pathologies.
Published on bit.bio's website, this article authored by Dr Tony Oosterveen dives into the close relationship between FTD and ALS with a focus on mechanisms underlying the neuropathology of both diseases.
On April 30th, 1939, baseball hall of famer Lou Gehrig stepped up to the plate with weary muscles. Over the course of Gehrig’s 15-year career, fans and teammates had dubbed him the “Ironhorse” as a nod to the fact that neither illness, injury, nor fatigue had ever caused him to miss a game. The man was, it seemed, invincible. But on this day, the Ironhorse’s 2130th consecutive game would also prove to be his last.1, 2

Though he was only 37, the motor neurons in Gehrig’s body had begun to deteriorate, weakening his muscles, degrading his coordination, and robbing him of his famed athleticism. Unbeknownst to him, these symptoms were the signs of amyotrophic lateral sclerosis (ALS), a rapidly progressive and fatal neurodegenerative condition that many now know as Lou Gehrig’s disease.
Gehrig’s declining health prevented him from playing another game, but his fame brought the world’s attention to this enigmatic disease and others like it.
In the sections that follow, we dive in on the latest research describing the underlying causes of ALS and a closely related condition known as frontotemporal dementia (FTD), two forms of neurodegenerative disease that share striking similarities in their molecular pathologies. We will also highlight the need for more robust, scalable, and consistent cell models of FTD and ALS.
What is amyotrophic lateral sclerosis (ALS)?
ALS is the most common motor neuron disease and is estimated to affect more than 80,000 people worldwide.3 All ALS patients share the same progressive dysfunction and death of motor neurons in either the primary motor cortex, the spinal cord, or both. This loss manifests as muscle paralysis and atrophy that can ultimately reduce a patient's ability to breathe. The average life expectancy following symptom onset is ~2-5 years.4, 5 And while patients share common molecular hallmarks, the wide heterogeneity among ALS subtypes suggests that it may not be a single disease, but rather a common outcome of multiple neurodegenerative processes.6
To this end, ALS patients often show overlapping clinical representation to other neurodegenerative disorders, particularly frontotemporal dementia, with particular similarities to the underlying molecular mechanisms of disease.
What is frontotemporal dementia (FTD)?
FTD is a neurodegenerative disease characterised by the progressive death of Glutamatergic and GABAergic neurons in the frontal and temporal lobes, with some evidence pointing to decreases in neurotransmitters like GABA contributing to disease presentation.7, 8 The cumulative loss of these neurons and the disruption of neurotransmitter levels can have a range of debilitating effects that alter patient behaviour and cognition. Similar to ALS, there appear to be numerous molecular factors contributing to the onset of FTD, which has led experts to suggest that FTD is not a singular disorder, but rather a shared neurodegenerative outcome resulting from various mechanisms.
Hallmarks of ALS and FTD
ALS and FTD are not always easy to distinguish, and disease symptoms show cross-over (Table 1). Roughly 50 percent of patients with ALS also develop cognitive or behavioural impairments, while 30 percent of FTD patients may also show motor neuron dysfunction.9 This overlap is mirrored at a genetic level, where current research suggests that approximately 20 percent of genes linked to one disease are also linked to the other.10 Given these similarities, many have begun to view these conditions as two points on a single neuropathological spectrum, with each driven by shared pathological processes.11
In this light, it’s not surprising that FTD and ALS present with many of the same molecular hallmarks, including atypical protein aggregates, lysosomal dysfunction, and axonal degeneration (among many others).7, 10-14 Studying these hallmarks and their relationship to disease pathology may not only advance our understanding of FTD and ALS, but uncover broader insights about the molecular mechanisms driving the function and potential dysfunction of the human brain.

Table 1. Similarities in the hallmarks of ALS and FTD[/caption]
Protein aggregates in ALS and FTD
Examination of neurons from both FTD and ALS patients reveals the existence of atypical protein aggregates in the cells’ cytoplasm. The specific proteins found in these aggregates can vary, but approximately 95 percent of ALS patients and 50 percent of FTD patients develop aggregates containing the TDP-43 protein (short for tar DNA binding protein 43).14
TDP-43 is a DNA and RNA binding protein that regulates the expression and splicing of several target transcripts.16 In healthy patients, nuclear expression of TDP-43 is ubiquitous throughout neurons in the central nervous system. In contrast, patients with ALS and FTD show a redistribution of TDP-43 in affected nerve cells, sometimes accompanied by a decrease in nuclear expression.17-19 Abnormal TDP-43 distribution coincides with increased TDP-43 phosphorylation as well as TDP-43 aggregation in the neuronal cytoplasm and axon. The cause of TDP-43 mislocalization is unclear.20, 21
Precisely how TDP-43 aggregates mediate neuronal toxicity is also not yet clear. There are several possibilities, but two main mechanisms highlighted in current research include toxicity stemming from the aggregates themselves, from a loss of TDP-43 nuclear activity, or a combination of the two among many other possibilities. Many potential toxic effects have been suggested, including the mis-splicing of the Stathmin-2 RNA transcript.14

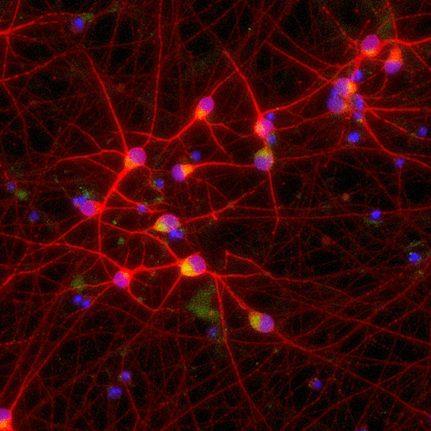
Image modified from Suk et al.
21Mislocalization of TDP-43 (red) causes various effects on neurons, ranging from disrupted RNA splicing to dysregulation of lysosomes and autophagic machinery. These effects promote neuron cell death and exacerbate ALS and FTD disease pathology. Targeting TDP-43 mislocalization is one of many potential therapeutic approaches.[/caption]
Stathmin-2 is one of many proteins that appear critical for the growth and stabilisation of axons, most likely by regulating the dynamics of axonal microtubules.22 Under normal conditions, TDP-43 prevents cells from using a cryptic splice site on the Stathmin-2 transcript. In ALS and FTD, however, when TDP-43 is sequestered in the cytoplasm, this cryptic splice site is de-repressed, enabling cells to access a cryptic poly-A site (normally hidden in an intron). Consequently, use of the site produces a transcript containing a premature poly-A tail that truncates the final protein. This truncated form of Stathmin-2 is unable to properly regulate microtubule dynamics, ultimately degrading axonal integrity.22
Axonal degeneration in ALS and FTD
Both ALS and FTD present with the pathological degradation of axons preceding cell death. Axons are supported by an underlying framework of protein filaments known as microtubules. The regulated growth and destruction of microtubules are essential to the growth of axons and their associated neurites. Additionally, microtubules serve as molecular highways along which proteins and other cargo can be ferried from the cell body to distal axonal locations.23
Destruction of the microtubule filaments can be catastrophic for neurons, leading to abnormal electrophysiology and eventual degeneration of the axon. Not surprisingly, diseases like FTD and ALS often feature abnormal regulation of microtubule associated proteins (MAPs) that influence microtubule dynamics.24, 25
Tau is another MAP that is frequently associated with FTD and can be found in atypical aggregates. One of the most well-studied MAPs, Tau is known to bind to growing microtubules where it helps to prevent the filament from unfurling.26 However, hyperphosphorylation of Tau can decrease its affinity for the microtubules and, in turn, the stability of axons.
Approximately 50 percent of FTD patients show abnormal regulation of Tau, whether it's through hyperphosphorylation or a change in the ratio of Tau isoforms that can lead to Tau aggregation.14 Either way, the end result is the same: microtubule stability goes down, and with it, axonal degeneration proceeds.
Lysosomal disorder in ALS and FTD
Recent studies suggest that FTD and ALS may be driven by disordered regulation and function of lysosomes.12 By sequestering and degrading cellular material, lysosomes play an invaluable role in maintaining cellular homeostasis. Loss of this function can result in a pathological build up of proteins and other substances that have a particularly pronounced effect on the central nervous system. Not surprisingly, lysosomal storage disorders often show CNS pathologies.
Several lines of evidence suggest the presence of lysosomal dysfunction in FTD and ALS. Tissues collected from affected patients, for example, show lipofuscin deposits, glycosphingolipids, and reduced nuclear localisation of transcription factor EB (TFEB)—a master transcriptional regulator of lysosomal genes. Additionally, many of the proteins associated with ALS and FTD, such as Tau and TDP-43, have been shown to disrupt lysosomal dynamics.12
Insights like these strongly suggest that whatever the trigger, lysosomal dysfunction can help fuel a catastrophic cascade that promotes protein aggregate formation, axonal collapse, and eventual cell death.
Towards understanding the neuropathological spectrum
Each of the hallmarks mentioned in Table 2 is common in FTD and ALS patients, but they are not unique to these diseases. The hallmarks are also variably observed in other neurodegenerative disorders, including Parkinson’s disease and Alzheimer’s disease.13 Therefore studying the relationship between protein aggregation, lysosomal dysfunction, and axonal degeneration can have implications well beyond FTD and ALS.

Table 2. Similarities in the cytology and molecular characteristics of ALS and FTD[/caption]
Current research suggests that these shared hallmarks have a causal relationship with one another, but the precise interplay between them is still being worked out. For example, TDP-43 mislocalization may alter the expression of key genes that regulate microtubule stability, but it may also influence lysosomal dynamics, autophagy, and mitochondrial function among many other processes. Whether TDP-43 mislocalization precedes lysosomal dysfunction and disease development, or vice versa, remains to be seen.
Researchers need accurate model systems that allow for the meticulous study of these molecular hallmarks to better understand (and hopefully treat) ALS, FTD, and other neurodegenerative diseases. While such a task has historically been difficult, new opportunities are opening up with the advancement of precision cellular reprogramming technology.
bit.bio has recently developed human iPSC-derived neurons that harbour a key mutation in the TARDBP gene encoding the TDP-43 protein and human iPSC-derived neurons with two different mutations in the MAPT gene encoding Tau. Preliminary data suggests that these cells show key hallmarks of FTD and ALS.
References
- “Gehrig Is Second on MLB’s Consecutive Games List.” MLB.com, www.mlb.com/news/most-consecutive-games-played-in-mlb-history-c282212708. Accessed 5 May 2023.
- April 30, 1939: Lou Gehrig Plays His Final Game with Yankees – Society for American Baseball Research. sabr.org/gamesproj/game/april-30-1939-lou-gehrig-plays-his-final-game-with-yankees/. Accessed 5 May 2023.
- Arthur, Karissa C., et al. “Projected Increase in Amyotrophic Lateral Sclerosis from 2015 to 2040.” Nature Communications, vol. 7, no. 1, 11 Aug. 2016, www.ncbi.nlm.nih.gov/pmc/articles/PMC4987527/, https://doi.org/10.1038/ncomms12408.
- Zarei, S., et al. “A comprehensive review of amyotrophic lateral sclerosis”. Surg Neurol Int. vol 16;6:171, Nov 2015, https://doi.org/10.4103/2152-7806.169561.
- Marin, B., et al. “Clinical and demographic factors and outcome of amyotrophic lateral sclerosis in relation to population ancestral origin”. Eur J Epidemiol vol. 31, 2016, pp. 229–245, https://doi.org/10.1007/s10654-015-0090-x
- “Frontotemporal Dementia vs. ALS.” ALS Therapy Development Institute, www.als.net/news/frontotemporal-dementia-vs-als/. Accessed 5 May 2023.
- Guo, Wenting, et al. “Current Advances and Limitations in Modeling ALS/FTD in a Dish Using Induced Pluripotent Stem Cells.” Frontiers in Neuroscience, vol. 11, 13 Dec. 2017, https://doi.org/10.3389/fnins.2017.00671.
- Huber, N., et al. “Deficient neurotransmitter systems and synaptic function in frontotemporal lobar degeneration-Insights into disease mechanisms and current therapeutic approaches”. Mol Psychiatry vol. 27, no. 3, Mar. 2022, pp.1300-1309. doi: https://doi.org/10.1038/s41380-021-01384-8.
- Abramzon, Yevgeniya A., et al. “The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia.” Frontiers in Neuroscience, vol. 14, 5 Feb. 2020, https://doi.org/10.3389/fnins.2020.00042.
- Liscic, Rajka Maria, et al. “From Basic Research to the Clinic: Innovative Therapies for ALS and FTD in the Pipeline.” Molecular Neurodegeneration, vol. 15, no. 1, 1 June 2020, https://doi.org/10.1186/s13024-020-00373-9.
- Ling, Shuo-Chien et al. “Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis.” Neuron vol. 79,3 (2013): 416-38. doi:10.1016/j.neuron.2013.07.033
- Root, Jessica, et al. “Lysosome Dysfunction as a Cause of Neurodegenerative Diseases: Lessons from Frontotemporal Dementia and Amyotrophic Lateral Sclerosis.” Neurobiology of Disease, vol. 154, July 2021, p. 105360, https://doi.org/10.1016/j.nbd.2021.105360.
- Virgilio, E., et al. “The Role of Tau beyond Alzheimer's Disease: A Narrative Review.” Biomedicines vol. 10 no. 4, 24 Mar. 2022, https://doi.org/10.3390/biomedicines10040760.
- Glass, Jonathan D. “Stathmin-2: Adding Another Piece to the Puzzle of TDP-43 Proteinopathies and Neurodegeneration.” Journal of Clinical Investigation, vol. 130, no. 11, 19 Oct. 2020, pp. 5677–5680, https://doi.org/10.1172/jci142854.
- Woollacott, Ione O. C., and Jonathan D. Rohrer. “The Clinical Spectrum of Sporadic and Familial Forms of Frontotemporal Dementia.” Journal of Neurochemistry, vol. 138, 15 June 2016, pp. 6–31, https://doi.org/10.1111/jnc.13654.
- Lee, Sebum, and Eric J. Huang. “Modeling ALS and FTD with IPSC-Derived Neurons.” Brain Research, vol. 1656, Feb. 2017, pp. 88–97, https://doi.org/10.1016/j.brainres.2015.10.003.
- Riva, N., et al. “Phosphorylated TDP-43 aggregates in peripheral motor nerves of patients with amyotrophic lateral sclerosis”. Brain vol. 145, no. 1, Mar. 29 2022, pp. 276-284. https://doi.org/10.1093/brain/awab285
- Giordana, M. T., et al. “TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis”. Brain Pathol vol. 20, no. 2, Mar. 2010, pp. 351-60. doi: https://doi.org/10.1111/j.1750-3639.2009.00284.x
- Neumann, M., et al. “Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis.” Science vol. 314. https://doi.org/10.1126/science.1134108
- Corcia, P., et al. “Phenotype and Genotype Analysis in Amyotrophic Lateral Sclerosis with TARDBP Gene Mutations.” Neurology, vol. 78, no. 19, 25 Apr. 2012, pp. 1519–1526, https://doi.org/10.1212/wnl.0b013e3182553c88.
- Suk, T.R., Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegeneration 15, 45 (2020). https://doi.org/10.1186/s13024-020-00397-1
- Melamed, Ze’ev, et al. “Premature Polyadenylation-Mediated Loss of Stathmin-2 Is a Hallmark of TDP-43-Dependent Neurodegeneration.” Nature Neuroscience, vol. 22, no. 2, 1 Feb. 2019, pp. 180–190, https://doi.org/10.1038/s41593-018-0293-z.
- Baas, Peter W., et al. “Stability Properties of Neuronal Microtubules.” Cytoskeleton, vol. 73, no. 9, Sept. 2016, pp. 442–460, https://doi.org/10.1002/cm.21286.
- Yang, Xiaoman, et al. “Common mechanisms underlying axonal transport deficits in neurodegenerative diseases: a mini review.” Front. Mol. Neurosci. vol 16, 24 April 2023, https://doi.org/10.3389/fnmol.2023.1172197
- Bodakuntla, Satish, et al. “Microtubule-Associated Proteins: Structuring the Cytoskeleton.” Trends Cell Bio. vol. 29, no. 10, Oct. 2019, pp. 804-819, https://doi.org/10.1016/j.tcb.2019.07.004
- Barbier, Pascale, et al. “Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects.” Front. Aging Neurosci., vol. 11, 7 Aug. 2019, https://doi.org/10.3389/fnagi.2019.00204
- Liguori, Francesco, et al. “Where and Why Modeling Amyotrophic Lateral Sclerosis.” International Journal of Molecular Sciences, vol. 22, no. 8, 12 Apr. 2021, p. 3977, https://doi.org/10.3390/ijms22083977
Author Bio:
 Dr Tony Oosterveen
Dr Tony Oosterveen
Dr Tony Oosterveen is a Principal Scientist and CNS lead at bit.bio, a company located in Cambridge, UK using proprietary technologies to robustly generate human cell types for research, drug discovery and cell therapy. During his academic career, he resolved a long-standing question in the field on how signalling molecules are interpreted at the transcriptional level to specify neuronal subtypes and developed a novel hESC-derived Parkinson’s disease model.