Exploring new drug targets in Alzheimer’s disease
Posted: 11 September 2020 | Monika Schmidt (University of Hradec Kralove), Sheraz Gul (Fraunhofer Institute) | No comments yet
Although many potential targets have been identified for Alzheimer’s disease (AD), there is no effective treatment for this debilitating condition. In this article, Monika Schmidt and Sheraz Gul delve into the key proteins implicated in AD and suggest how phenotypic assays could aid in AD drug discovery.
There is no effective treatment for this debilitating disease”
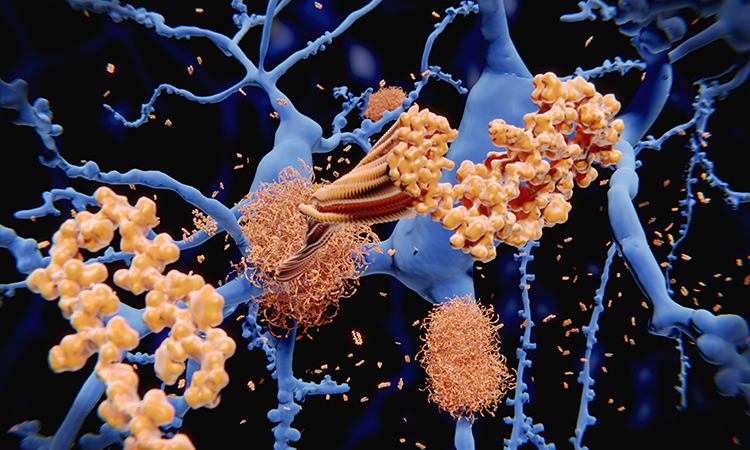
DEMENTIA AND Alzheimer’s disease (AD) are terms describing symptoms associated with cognitive impairment in humans. Currently, 40-50 million people across the globe are living with some form of dementia, a number which has doubled since 1990. Women are more likely to suffer from dementia and it was the fifth leading cause of death in 2016 globally.1 One reason for the increase in numbers of people suffering from dementia is the lack of a therapeutic that can cure or slow the disease. Thus far, more than 200 proteins or enzymes have been implicated in AD with many hypotheses linking them with the disease, but despite extensive AD research, its molecular mechanism remains largely unknown. This is in part because no single factor has been identified as a causative agent. However, it does involve neuronal cell death which is linked to deposition of amyloid beta (Aß) peptides followed by neuro-inflammation, neuronal death and eventually impairment of cognitive functions.
In general, two major forms of AD are recognised: the minor form, which has an early-onset (familial Alzheimer’s disease [FAD]) and is estimated to be the cause of less than five percent of all AD cases; and a late-onset, sporadic form, which causes over 95 percent of AD. In the case of FAD, neurological symptoms appear in early middle age, and the accelerated onset is due to the presence of genetic mutations altering the cleavage of amyloid precursor protein (APP), eventually leading to the pathogenic Aß peptide.2 These APP mutations are located near their cleavage sites, which affects both the Aß peptide production itself and the ratio between Aß40 and Aß42. The trigger mechanism that leads to Aß increase in late-onset sporadic AD remains unknown.3
The major AD proteins – Aß and tau
A major drawback of AD research is the absence of suitable animal models that are representative
of the human disease at the molecular level”
Aß is the cleavage peptide of APP. To create Aß, APP is processed by beta-amyloid cleaving enzyme (BACE-1) at its amino terminus, then cleaved at its carboxyl terminus by ß-secretase. This Aß peptide is released extracellularly and undergoes rapid degradation in healthy brains. However, in AD, dysregulation and accumulation of Aß peptide causes neuronal toxicity.4 The conditions that lead to this pathological behaviour of the Aß peptide are largely unknown.
The second major protein involved in the pathological hallmarks of AD is tau protein, a microtubule-associated protein, which under normal physiological conditions is important for axonal transport and thus is critical for neuronal functions. Under pathological conditions, tau protein forms aggregated, phosphorylated tangles within cells. This pathological alteration of tau protein, known as tauopathy, is a shared hallmark of some neurological disorders such as AD, Parkinson´s disease and frontotemporal dementia. Hyperphoshorylation of tau protein followed by its dissociation from axonal microtubules and formation of abnormal neurofibrillary tangles disrupts cell transport mechanisms and can lead to neuronal degeneration, cell toxicity and cell death.5,6
Aß protein hypothesis in AD
The Aß protein cascade hypothesis was established in the early 1990s.7,8 The main hallmark of this widely accepted AD hypothesis is that Aß peptides of different lengths – from 39 to 43 amino acid residues – accumulate to create extracellular senile plaques and these amyloid deposits are the trigger of the neuropathology. Aß peptide monomers, predominantly of 40 and 42 amino acid residues long, are very hydrophobic and tend to self-aggregate with a characteristic β-sheet secondary structure and reverse turn supersecondary structures. These monomers form the protofibrils and fibrils that are deposited in the brain as pathological extracellular plaques.
 The importance of the Aß protein cascade hypothesis was supported by the discovery of several mutations in genes of APP and presenilins, which are catalytic components of ß-secretase, leading to generation of the amyloidogenic form of Aß peptide, namely the Aß42 isoform. This hypothesis was also based on the duplication of the APP gene on chromosome 21 in Down´s syndrome or individuals having microduplication of sub-regions of chromosome 21, who typically present AD in their mid-50s.9 Moreover, this hypothesis is also supported by the discovery of AD protective mutations leading to the generation of Aß peptide that has altered aggregation properties. Thus, carriers of this mutation have a lower risk of clinical AD.10 This contrasts with genetically inherited FAD, where mutations in APP and presenilins lead to an elevated Aß peptide and/or Aß42/Aß40 ratio, whilst the causative agent of Aß dyshomeostasis in late-onset, sporadic form is still unknown.
The importance of the Aß protein cascade hypothesis was supported by the discovery of several mutations in genes of APP and presenilins, which are catalytic components of ß-secretase, leading to generation of the amyloidogenic form of Aß peptide, namely the Aß42 isoform. This hypothesis was also based on the duplication of the APP gene on chromosome 21 in Down´s syndrome or individuals having microduplication of sub-regions of chromosome 21, who typically present AD in their mid-50s.9 Moreover, this hypothesis is also supported by the discovery of AD protective mutations leading to the generation of Aß peptide that has altered aggregation properties. Thus, carriers of this mutation have a lower risk of clinical AD.10 This contrasts with genetically inherited FAD, where mutations in APP and presenilins lead to an elevated Aß peptide and/or Aß42/Aß40 ratio, whilst the causative agent of Aß dyshomeostasis in late-onset, sporadic form is still unknown.
Availability of model systems for AD
A major drawback of AD research is the absence of suitable animal models that are representative of the human disease at the molecular level. The transgenic knock-in mice models currently in use do not reflect all the disease conditions in the human brain and the plaques and tangles that develop in the mouse brain are different. In addition, the plaques developing in transgenic mice models appear at a relatively early age and there should be the discrepancy between a non-aged and aged brain. In 2014, a human neural cell culture model of AD was reported that developed amyloid and tau pathology. This three-dimensional (3D) model has an elevated Aß40 and Aß42 phenotype together with increased Aß42/Aß40 ratio. Moreover, the 3D model also exhibited elevated endogenous protein tau aggregates11 as a consequence of direct Aß tauopathy induction.
Phenotypic assays in AD drug discovery
 Although many potential targets have been implicated in AD, there is no effective treatment for this debilitating disease. However, there are several marketed drugs for the dementia phase of AD, namely donepezil, rivastigmine and galantamine, which are cholinesterase inhibitors, and memantine, which is an antagonist to the N-methy-lD-aspartate receptor. All the currently used drugs are for the treatment of symptomatic patients with mild to moderate AD and they do not target the root cause of the disease.12 As the AD phenotype is well characterised, the use of complex cell-based assays that recapitulate the disease, such as 3D-model systems,11 are compatible with screening compound libraries, albeit in low throughput. Appropriate endpoints in these cell-based assays can be the measurement and quantification of Aß42 and Aß40 levels when exposing cells to compounds. Subsequent evaluation of compounds that yield the appropriate phenotype can subsequently be evaluated in target-based assays to determine their mode-of-action or explored in an in vivo setting if they have suitable properties.
Although many potential targets have been implicated in AD, there is no effective treatment for this debilitating disease. However, there are several marketed drugs for the dementia phase of AD, namely donepezil, rivastigmine and galantamine, which are cholinesterase inhibitors, and memantine, which is an antagonist to the N-methy-lD-aspartate receptor. All the currently used drugs are for the treatment of symptomatic patients with mild to moderate AD and they do not target the root cause of the disease.12 As the AD phenotype is well characterised, the use of complex cell-based assays that recapitulate the disease, such as 3D-model systems,11 are compatible with screening compound libraries, albeit in low throughput. Appropriate endpoints in these cell-based assays can be the measurement and quantification of Aß42 and Aß40 levels when exposing cells to compounds. Subsequent evaluation of compounds that yield the appropriate phenotype can subsequently be evaluated in target-based assays to determine their mode-of-action or explored in an in vivo setting if they have suitable properties.
About the author
 Sheraz Gul is the Head of Assay Development and Screening at the Fraunhofer Institute, Germany. He has professional experience in the field of drug discovery, assay development and screening gained whilst employed in academia. He has co-authored 80 peer-reviewed papers, book chapters and patents including the Enzyme Assays: Essential Data handbook. He also has an interest in education and thus far has organised 46 drug discovery workshops since 2011 across the globe and trained 880 scientists.
Sheraz Gul is the Head of Assay Development and Screening at the Fraunhofer Institute, Germany. He has professional experience in the field of drug discovery, assay development and screening gained whilst employed in academia. He has co-authored 80 peer-reviewed papers, book chapters and patents including the Enzyme Assays: Essential Data handbook. He also has an interest in education and thus far has organised 46 drug discovery workshops since 2011 across the globe and trained 880 scientists.
Monika Schmidt is currently a researcher at Faculty of Science, University of Hradec Kralove in the Czech Republic. Her team is working on the development of new drugs against AD and she is focusing on mitochondrial targets involved in AD. Her work is based on recombinant enzyme cloning and preparation and subsequent biochemical testing in vitro.
References
- Nichols, Emma, et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. The Lancet Neurology 18.1 (2019): 88-106.
- Barber, Robert C. The genetics of Alzheimer’s disease. Scientifica 2012 (2012).
- Holtzman, David M., John C. Morris, and Alison M. Goate. Alzheimer’s disease: the challenge of the second century. Science translational medicine 3.77 (2011): 77sr1-77sr1.
- Kametani, Fuyuki, and Masato Hasegawa. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Frontiers in neuroscience 12 (2018): 25.
- Khan, Shahzad S., and George S. Bloom. Tau: the center of a signaling nexus in Alzheimer’s disease. Frontiers in Neuroscience 10 (2016): 31.
- Reddy, P. Hemachandra. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain research 1415 (2011): 136.
- Selkoe, Dennis J. The molecular pathology of Alzheimer’s disease. Neuron 6.4 (1991): 487.
- Hardy, John A., and Gerald A. Higgins. Alzheimer’s disease: the amyloid cascade hypothesis. Science 256.5054 (1992): 184.
- Rovelet-Lecrux, Anne, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nature genetics 38.1 (2006): 24.
- Kero, Mia, et al. Amyloid precursor protein (APP) A673T mutation in the elderly Finnish population. Neurobiology of aging 34.5 (2013): 1518-e1.
- Choi, Se Hoon, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature, 2014, 515.
- Atri, Alireza. “Current and Future Treatments in Alzheimer’s Disease.” Seminars in neurology. Vol. 39. No. 02. Thieme Medical Publishers, 2019.
Related topics
Assays, Drug Development, Drug Targets, Neurosciences, Targets
Related conditions
Alzheimer's disease (AD), Dementia, Down’s syndrome
Related organisations
Horizon