Tracking SARS-CoV-2 with genome sequencing
Posted: 2 December 2020 | Nikki Withers (Drug Target Review) | No comments yet
Researchers have been tracking SARS-CoV-2 by sequencing the genomes of virus samples collected from diagnostic testing. They hope that using next-generation sequencing (NGS) on SARS-CoV-2 will help to accurately diagnose the novel coronavirus, identify mutations and track its history. This article explores the findings of their latest study and what this means for future research.
COVID-19 is caused by infection with Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and has been responsible for over 200,000 deaths in the US alone. The first reported SARS‑CoV-2 clusters appeared in the Wuhan province of China during December 2019 and it has since rapidly spread across the world. Social distancing, local and national lockdowns and quarantine of infected persons have proven successful in limiting the impact of COVID-19. However, for these public health measures to remain effective and sustainable, it is important to understand the pathways of transmission through contact tracing and virus testing.
To investigate this further, a team of researchers in the US has been using NGS to track mutations in the SARS-CoV-2 virus, which they hope could help with transmission tracing, diagnostic-testing accuracy and vaccine effectiveness. “Once you have the virus’ genetic sequence with NGS then you can start asking more questions,” said Dirk Dittmer, Professor of Microbiology and Immunology at the UNC School of Medicine in North Carolina and senior author of the study.1 “Where have we seen this exact sequence before? Did it come from a different state or country? When did this patient travel there and who else may have it?”
According to Dittmer, this type of virus monitoring is also important in diagnostic testing. He explained that much of the testing developed to diagnose COVID-19 looks for one portion of the gene sequence that causes the novel coronavirus. However, if that sequence mutates, the test is no longer accurate and results will be affected. Within their study, which was published in Cell Reports,1 Dittmer’s team found variations in the virus’ genetic sequence, but reported that none were located in the portion of the virus targeted in common diagnostic testing.
NGS analysis
Once you have the virus’ genetic sequence with NGS then you can start asking more questions”
While prior studies have focused on high case-density locations in the US, such as the northern and western metropolitan areas, this study is the largest to focus on suburban and rural communities, according to the US researchers. The team was able to reconstruct the mutational landscape of cases seen at the UNC Medical Center, a tertiary clinical care centre in North Carolina. From 30 March through to 8 May 2020, 175 samples from confirmed COVID-19-positive patients were analysed.
Of the samples tested, 57 percent carried the Spike (S) protein D614G single-nucleotide variant (SNV), a mutation implicated in higher pathogenicity of the virus. The presence of this variant is associated with a higher genome copy number and its prevalence has expanded throughout the pandemic. The researchers said that the genetic variations found in these samples also support the hypothesis that the majority of cases in North Carolina originated from people travelling within the US rather than internationally. Of note, while the current study was under review, many other studies confirmed the importance of the D614G SNV and its biological and clinical properties.
One large deletion was identified in four independent samples – 14 nucleotides were deleted beginning at position 29745. This region is within the previously recognised “coronavirus 3′ stem‑loop II-like motif (s2m)”. To confirm their deep-sequencing results, the researchers performed 3′ UTR site-specific amplification and Sanger-based sequencing and found that the variant 3′ end does not destroy overall folding but introduces a shorter stable hairpin (Figure 1). How this mutation affects viral fitness remains to be established, they said.
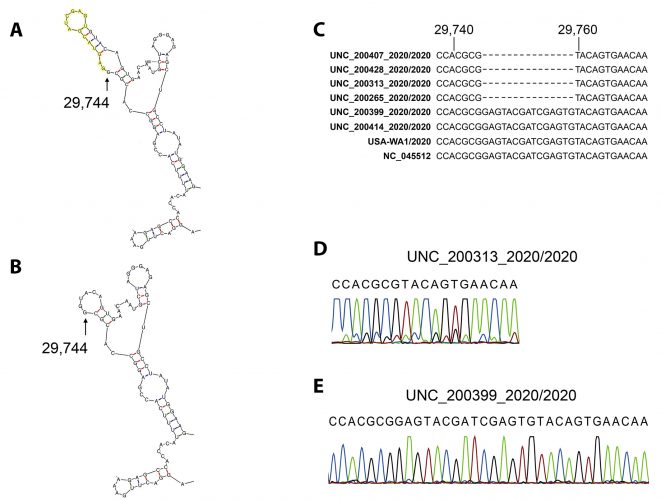
Figure 1: A) Predominant Mfold prediction of the 3′ end of NC_045512 with deletion bases indicated in yellow; B) Predominant Mfold prediction of the 3′ end of NC_045512 delta14; C) Sequence alignment of 3′ UTR deletion mutants with other representative SARS-CoV-2 isolates; D) Sanger sequencing confirmation
of the 3′ UTR deletion mutant UNC_200313_2020/2020; E) Sanger sequencing confirmation wild-type sequence for isolate UNC_200399_2020/2020. Image credit: Adapted from McNamara R, Caro-Vegas C, Landis J, Moorad R, Pluta L, Eason A, et al.1
Conclusions and future work
Concluding their study, the team confirmed that they had generated exhaustive SNV information representing the introduction and spread of SARS-CoV-2 across a suburban low-density area in the southern US. “All samples were from symptomatic cases and the majority of genomes clustered with variants that predominate the outbreak in the US, rather than Europe or China,” they said. “This supports the notion that the majority of US cases were generated by domestic transmission.”
Changes in other areas of the genetic sequence can not only disrupt testing but hinder the effectiveness of vaccines”
Dittmer noted that they are still concerned about future mutations. “It is inherent in a virus’ nature to mutate. Changes in other areas of the genetic sequence can not only disrupt testing but hinder the effectiveness of vaccines.” This is why Dittmer’s lab has been collaborating with multiple other labs at UNC-Chapel Hill to stay up to date on what, if any, changes should be made to testing protocols and possible vaccine development. They are receiving positive SARS-CoV-2 samples from the lab of Melissa Miller, Director of UNC Medical Center Microbiology and Molecular Microbiology Laboratories, where UNC’s COVID-19 diagnostic testing was developed and put in place on 16 March. “Because we are only looking at one gene sequence for the virus, we have told the US Food and Drug Administration (FDA) that we will continually monitor for changes in this gene sequence so that we can be assured that our test is still reliable,” said Miller, a co-author of the study. “NGS will help us do that.”
With a grant from the North Carolina Policy Collaboratory based at UNC-Chapel Hill, Dittmer’s lab will continue using NGS to track the SARS-CoV-2 virus through the remainder of 2020. The goal is to enrol every patient at UNC Hospitals with flu or respiratory symptoms for COVID-19 diagnostic testing. These samples will be sequenced and compiled to form a comprehensive profile of any virus that these patients carry, providing information that will continue to help researchers in their fight against SARS-CoV-2 and potentially other novel coronaviruses.
Acknowledgement
This work was funded by public health service grants CA016086, CA019014 and CA239583 to DPD. Funding was also provided by the University Cancer Research Fund and the UNC School of Medicine.
About the author
Nikki Withers is the Editor of Drug Target Review and European Pharmaceutical Review.
Reference
- McNamara R, CaroVegas C, Landis J, Moorad R, Pluta L, Eason A, et al. HighDensity Amplicon Sequencing Identifies Community Spread and Ongoing Evolution of SARS-CoV-2 in the Southern United States. Cell Reports. 2020;33(5):108352.
Related topics
Disease Research, DNA, Genetic Analysis, Genomics, Next-Generation Sequencing (NGS), Research & Development, Sequencing
Related conditions
Covid-19
Related organisations
North Carolina Policy Collaboratory, University of North Carolina (UNC), University of North Carolina at Chapel Hill (UNC-Chapel Hill), US Food and Drug Administration (FDA)
Related people
Melissa Miller, Professor Dirk Dittmer