Covalent fragment screening in cell-based phenotypic models of disease: a collaborative approach
Posted: 22 June 2021 | Dr Andrew Powell (GlaxoSmithKline), Dr David House (GlaxoSmithKline), Dr Jacob Bush (GlaxoSmithKline), Dr Katrin Rittinger (The Francis Crick Institute), Dr Simon Boulton (The Francis Crick Institute) | No comments yet
The application of chemical perturbation approaches in phenotypic models is often used to identify protein targets for therapeutic discovery. Increasingly, small molecule fragments which covalently bind to their protein targets are being used to explore the druggable proteome and the resulting fragment‑protein interactions are characterised by chemoproteomic techniques. In this article, researchers from the Francis Crick Institute (Crick) and GlaxoSmithKline (GSK) outline a new collaborative initiative with the aim to establish a Systems Chemical Biology platform that enables covalent fragment library screening in phenotypic models of human disease, along with chemoproteomic annotation of the fragment library members to enable rapid tool molecule generation for tractable targets.
Target selection and small molecule screens
The identification of tractable protein targets for efficacious therapeutic intervention is one of the most significant challenges in the discovery of novel medicines. Evidence that therapeutics which target proteins with strong genetic linkage to disease are more likely to be successful in the clinic1,2 has accelerated investment in genome‑wide association studies (GWAS) and large‑scale computational analyses. Genetic perturbation studies (eg, siRNA, shRNA or CRISPR‑Cas9 gene knockdown) in phenotypic models of disease provide further opportunity to link gene products to disease pathways.3 Ultimately, however, the targets identified through these studies need to prove tractable for modulation by a therapeutic molecule.
In the case of small molecule drugs, protein targets require pockets for the molecule to bind to with sufficient affinity to mediate a modulatory effect on function and deliver therapeutic benefit. Evidence of a protein’s small molecule tractability is often inferred from drug discovery experience with similar proteins, but where previously ‘undrugged’ protein classes are implicated by genetic studies there is often insufficient weight of data to classify the protein’s tractability. In these cases, small molecule screens would be undertaken with lower confidence in a successful outcome.
Target-specific screens are configured to identify novel ligands from small molecule libraries. Assay formats depend on protein class, but could include measures of enzyme activity, signalling pathway activation/inhibition, competition with a probe ligand or a measure of small molecule‑target binding affinity.4-7 Hit molecules are triaged through a cascade of additional assays to identify leads with target selectivity and to confirm the molecules’ impact on disease-associated pathways in phenotypically relevant models.
An alternative approach to identify novel small molecules as leads for novel therapeutic development is phenotypic drug discovery. In this scenario, small molecule libraries are screened in assays that closely recapitulate the biological pathways believed to drive the disease.8 The hits identified may modulate any druggable proteins involved in driving the biological phenotype, so the approach is not restricted to a specific target‑disease hypothesis and can be considered ‘target agnostic’. When efficacious lead molecules are identified, research is conducted to determine the mechanism through which the molecules mediate their impact. Approaches may include analysis of disease‑associated biomarkers and canonical signalling pathways (through transcriptomics, proteomics and metabolomics), however chemoproteomics studies are often required to determine with which protein(s) the lead molecule(s) are interacting.9,10
Covalent fragment screening
Where a target can be expressed and purified, structural and biophysical techniques can be used to determine whether small molecule binding pockets exist in the protein’s structure. Fragment-based screening has proved a powerful means of identifying small molecule ligands for these binding pockets.11-14 Fragments are low complexity compounds that can be used to efficiently sample chemical space to identify novel ligands for binding pockets. Fragment-protein binding affinities are relatively weak (10-4-10-2 M), so the resulting ligands require further optimisation to increase affinity and this limits their utility for deployment in cell‑based phenotypic screens.
The combination of covalent fragment screening and cell-based chemoproteomic profiling in phenotypic models will drive a paradigm change”
However, fragment compounds can be generated with reactive functional groups to enable the weak, reversible protein‑ligand interaction to be captured through the formation of a covalent bond.15-17 The reactive groups form covalent bonds with proximal, nucleophilic amino acids and reactivity can be tailored to distinct amino acids by judicious choice of electrophile. Covalent fragments can therefore exhibit increased potency over low affinity fragment-protein interactions and do so in a time-dependent manner.
Covalent fragment screening has been applied through affinity-based screens against readily purified proteins (Figure 1A). In these studies, libraries of covalent fragments are incubated with the protein and any fragments that covalently bind are identified through a mass shift detected by intact liquid chromatography‑mass spectrometry (LC-MS). This approach has identified novel covalent probes for previously intractable proteins KRas and HOIP.18,19 Recently, advances in mass spectrometry‑based proteomic analysis have increased sensitivity and throughput to enable covalent fragment screens using cell lysates or live cells.20 In this context, covalent fragments are identified which bind target proteins in their native state, complete with post‑translational modifications and/or in regulatory complexes. These activity‑based protein profiling (ABPP) techniques are configured using reactive probes which can be conjugated to affinity tags to enrich the covalently-bound proteins for LC‑MS/MS analysis.21 Covalent fragments binding proximal to the targeted nucleophilic amino acid residue compete with the reactive probe, therefore fragment bound proteins are depleted in the LC‑MS/MS analysis (competitive ABPP, Figure 1B).20,22 Alternatively, covalent fragments which have been functionalised through the addition of a “clickable” group (eg, an alkyne) can be exploited and the interacting proteins directly enriched for LC‑MS/MS analysis by bio‑orthogonal ligation of an affinity tag (eg, by copper-catalysed azide‑alkyne cycloaddition).
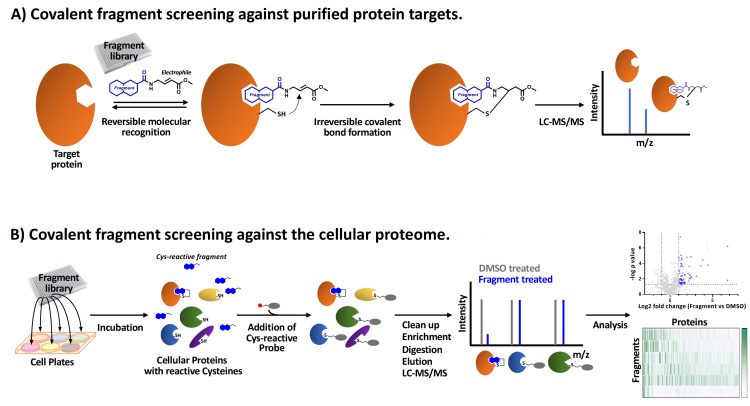
Figure 1: Covalent fragment screening approaches. A) Purified protein incubated with covalent fragment library members. A cysteine reactive fragment with an acrylate ester electrophile is shown as an example. Fragment-mediated reversible molecular recognition of a pocket in proximity to a reactive cysteine enables a covalent bond to form. Covalently bound fragments are identified by LC-MS/MS. B) Covalent fragment library members are incubated in cells and bind to proteins with complementary binding pockets and proximal reactive amino acids (eg, cysteine). A reactive probe molecule linked to an affinity-tag (eg, iodoacetamide desthiobiotin) is used to enrich proteins not covalently bound by fragments. Following digestion and elution, proteins are identified by LC-MS/MS and compared to control DMSO-treated cell samples to identify fragments which compete with the reactive probe (competitive ABPP). Analysis generates a matrix of covalent fragment-protein interaction data.
In a landmark study,22 a pilot library was used to probe the ligandable ‘cysteinome’ of two cancer cell proteomes. Of 637 unique liganded proteins, 545 (86 percent) were non-DrugBank proteins and, significantly, many of these were targets such as transcription factors and adaptor proteins, which have classically been regarded as intractable to small molecule perturbation. Further studies23-26 have coupled covalent fragment screening with chemoproteomics to directly map small molecule‑protein interactions in live cells and to investigate reactivity changes across the proteome in different activation states. These studies demonstrate the potential to carry out covalent fragment screens in in vitro phenotypic models of disease thereby enabling target-agnostic probing of biological systems and ultimately to relate phenotypic effects to distinct ligandable targets. Where previously fragment‑based screening was conducted against one protein at a time, cell-based covalent fragment screens represent a step change by accessing multiple proteins simultaneously and removing the need for protein purification for hit identification. Although most research to investigate the application of covalent fragments in cell-based proteomic profiling has exploited fragments with cysteine-reactive electrophiles, fragments with targeted reactivity to other amino acids are also being investigated.23,27 These studies will expand the ligandable proteome accessible to covalent fragment approaches.
Target identification – cell-based phenotypic screens of covalent fragment libraries
Research developing the use of chemoproteomic‑based covalent fragment screens to study biology has emerged recently.24,25,28 Mass spectrometry-based analysis now enables the chemoproteomic characterisation of covalent fragment reactivity in cell-based disease models, such that the potential targets of biologically active covalent fragments can be identified. Recently, the Crick and GSK secured Prosperity Partnership funding from the UK Engineering and Physical Sciences Research Council to develop a chemical biology platform capable of simultaneously identifying disease targets and functional chemical probes. The goal of this project, led by Simon Boulton (Crick) and David House (GSK), is to optimise covalent fragment library design and chemoproteomic workflows and to deploy covalent fragment screening in cell‑based phenotypic assays, thereby generating rich datasets describing the biological fingerprints of each fragment (Figure 2). Expanding the repertoire of covalent fragment-targeting chemistries will capture more ligand-protein interactions than cysteine‑reactive libraries alone. Due to their low complexity, each covalent fragment is likely to bind to multiple proteins. Integrating the fragment chemotype, chemoproteomic and biological fingerprint datasets will enable the application of machine learning analytics to identify opportunities to further evolve the covalent fragment libraries, for example, to increase proteome coverage and to identify covalent fragments which link ligandable protein targets to physiologically-relevant phenotypes. These covalent fragment tools can then be optimised to modulate their reactivity, increase selectivity and potentially be used to configure competition experiments to identify alternative ligands (covalent or non-covalent) with higher affinity.
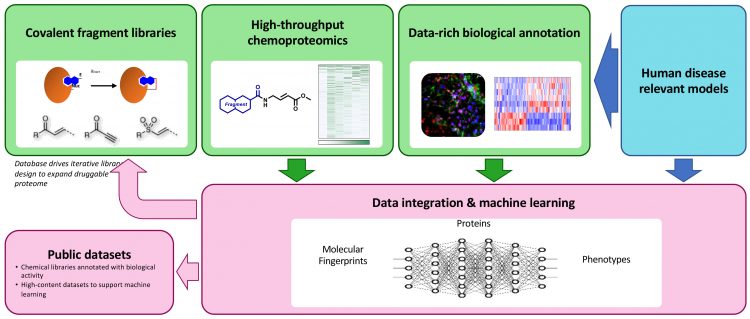
Figure 2: Overview of the EPSRC-funded Crick-GSK Prosperity Partnership Systems Chemical Biology Project
In developing the technology through collaboration, the project will benefit from the combined expertise of the partner organisations, including deep understanding of specific biological pathways, in vitro biological assays for disease‑relevant phenotypes, chemical biology, cell‑based chemoproteomic profiling and high‑content imaging capabilities along with experience in complex data integration and analysis. The impact of the technology will be explored in a range of disease models, from cancer and complex cell signalling models to infection and host immune response models.
Data integration and analysis
Cell-based chemoproteomic profiling will generate data describing the reactivities of each fragment within the library and enable the rapid selection of fragments to study specific targets. The Crick and GSK project aims to develop optimised and standardised data pipelines to extract and transform the data for uploading to a central database. The data will be annotated with globally recognised chemical and biological ontologies and robust pipelines will be established for data loading. Once developed, the database and associated unprocessed datasets will be accessible to the broader scientific community.
Fragment-based screening has proved a powerful means of identifying small molecule ligands”
Machine learning analysis will drive the expansion of the covalent fragment libraries to increase biological diversity (coverage of the proteome and phenotypes) and identify proteins that offer potential for therapeutic intervention in each disease model, as well as the tool molecules to further study these associations. Machine learning algorithms will be trained on full matrix chemoproteomic data and paired with active learning approaches to steer new library compounds towards those proteins which are under-represented in the dataset. Categorisation and clustering of high-content data, alongside reference to gene perturbation datasets, will provide the foundations for iterative library growth to expand the coverage of biological space and elucidate known and novel phenotypes.
Summary
The combination of covalent fragment screening and cell-based chemoproteomic profiling in phenotypic models will drive a paradigm change from a gene-centric view of targets and disease mechanisms to a spatially and functionally resolved view, providing complementarity to genetic screening approaches. In contrast to genetic perturbation studies, the identification of targets through this chemical perturbation route will yield proteins with binding pockets amenable to small molecule drug discovery. Ultimately, the combination of chemical tractability, along with genetic-disease association, will help to accelerate the discovery and development of novel medicines for patients.
About the authors
 Dr Andrew Powell is a group leader within GSK’s Chemical Biology department and Biology Director for the Crick-GSK LinkLabs collaboration. He obtained his degree in microbiology from Queen Mary College (University of London) and MSc in applied molecular biology and biotechnology from University College London. Andrew completed his PhD in cell biology and biochemistry at the Ludwig institute for Cancer Research, UCL branch. He joined GSK in 1996 to develop cellular assays for early discovery research and has led projects to discover novel ion channel lead molecules.
Dr Andrew Powell is a group leader within GSK’s Chemical Biology department and Biology Director for the Crick-GSK LinkLabs collaboration. He obtained his degree in microbiology from Queen Mary College (University of London) and MSc in applied molecular biology and biotechnology from University College London. Andrew completed his PhD in cell biology and biochemistry at the Ludwig institute for Cancer Research, UCL branch. He joined GSK in 1996 to develop cellular assays for early discovery research and has led projects to discover novel ion channel lead molecules.
 Dr Jacob Bush is a group leader in the Chemical Biology department at GSK. After completing a degree in chemistry from the University of Oxford, Jake studied for a DPhil in chemical biology in the lab of Chris Schofield. With an interest for translational science, he joined GSK in 2015 to explore ways to accelerate drug discovery. By combining chemoproteomic and chemogenomic approaches using covalent fragments, Jake and his team aim to discover new targets with therapeutic potential and use machine learning algorithms to accelerate the drug discovery process.
Dr Jacob Bush is a group leader in the Chemical Biology department at GSK. After completing a degree in chemistry from the University of Oxford, Jake studied for a DPhil in chemical biology in the lab of Chris Schofield. With an interest for translational science, he joined GSK in 2015 to explore ways to accelerate drug discovery. By combining chemoproteomic and chemogenomic approaches using covalent fragments, Jake and his team aim to discover new targets with therapeutic potential and use machine learning algorithms to accelerate the drug discovery process.
 Dr David House is UK Head of Chemical Biology at GSK. After obtaining a degree in Natural Sciences from the University of Cambridge, he completed his PhD in asymmetric synthesis followed by chemistry postdocs at the University of Geneva and Oxford. He joined GSK in 2003 as a medicinal chemist and has led projects from an early stage up to the point of clinical candidate selection. In 2015 David became part of a small team of scientists which established the Crick‑GSK LinkLabs, a pre‑competitive chemical biology collaboration focussed on technologies for target identification and validation, which he now heads.
Dr David House is UK Head of Chemical Biology at GSK. After obtaining a degree in Natural Sciences from the University of Cambridge, he completed his PhD in asymmetric synthesis followed by chemistry postdocs at the University of Geneva and Oxford. He joined GSK in 2003 as a medicinal chemist and has led projects from an early stage up to the point of clinical candidate selection. In 2015 David became part of a small team of scientists which established the Crick‑GSK LinkLabs, a pre‑competitive chemical biology collaboration focussed on technologies for target identification and validation, which he now heads.

Dr Katrin Rittinger is a Senior Group Leader at the Francis Crick Institute. After obtaining a degree in chemistry from the University of Heidelberg she completed her PhD at the Max Planck Institute for Medical Research in Heidelberg followed by a postdoc at the MRC-National Institute for Medical Research in London. She established her independent group in 2000 at the MRC‑NIMR, which became part of the Francis Crick Institute in 2015. Her group is studying protein ubiquitination as a key regulatory mechanism to drive cellular behaviour and is applying structural and biochemical approaches to understand the regulation and molecular mechanisms of enzymes catalysing protein ubiquitination. In parallel, she is collaborating with the Crick-GSK LinkLabs to develop chemical probes that target the ubiquitin system.
 Dr Simon Boulton is a Senior Group Leader and Ambassador for Translation at the Francis Crick Institute. Simon’s academic work has led to the discovery of novel DNA repair genes and provided molecular insights into genome instability disorders and cancer. Simon is also co-founder and VP Science Strategy at Artios Pharma Ltd, a biotech company that is developing DNA repair inhibitors to selectively kill cancer cells. Simon is an EMBO Member, Fellow of the Academic of Medical Sciences and has been awarded the Royal Society Francis Crick Medal, the EMBO Gold Medal and the Paul Marks Prize for Cancer Research.
Dr Simon Boulton is a Senior Group Leader and Ambassador for Translation at the Francis Crick Institute. Simon’s academic work has led to the discovery of novel DNA repair genes and provided molecular insights into genome instability disorders and cancer. Simon is also co-founder and VP Science Strategy at Artios Pharma Ltd, a biotech company that is developing DNA repair inhibitors to selectively kill cancer cells. Simon is an EMBO Member, Fellow of the Academic of Medical Sciences and has been awarded the Royal Society Francis Crick Medal, the EMBO Gold Medal and the Paul Marks Prize for Cancer Research.
References
- Nelson MR, Tipney H, Painter JL, et al. (2015). The support of human genetic evidence for approved drug indications. Nature Genetics. 47(8):856-860.
- King EA, Davis JW, Degner JF (2019) Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 15(12):e1008489.
- le Sage C, Lawo S, Cross BCS (2020). CRISPR: A Screener’s Guide. SLAS Discovery. 25(3):233-240.
- Macarron R, Banks M, Bojanic D, et al. (2011). Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov. 10:188-195.
- https://www.drugtargetreview.com/article/61883/high-throughput-screening-as-a-method-fordiscovering-new-drugs/
- Leveridge M, Chung C-W, Gross JW, et al. (2018). Integration of Lead Discovery Tactics and the Evolution of the Lead Discovery. SLAS Discovery. 23(9):881-897.
- Wigglesworth MJ, Murray DC, Blackett CJ, et al. (2015). Increasing the delivery of next generation therapeutics from high throughput screening libraries. Curr Opin Chem Biol. 26:104-110.
- Moffat, JG, Vincent F, Lee JA. (2017). Opportunities and Challenges in Phenotypic Drug Discovery: An Industry Perspective. Nat. Rev. Drug Discov. 16:531-543.
- Chung C, Coste H, White JH (2011). Discovery and Characterization of Small Molecule Inhibitors of the BET Family Bromodomains. J. Med. Chem. 54:3827-3838.
- Ketley A, Wojciechowska M, Ghidelli-Disse S, et al. (2020). CDK12 inhibition reduces abnormalities in cells from patients with myotonic dystrophy and in a mouse model. Sci. Transl. Med. 12(541):eaaz2415.
- Leach AR, Hann MM (2011). Molecular complexity and fragment-based drug discovery: ten years on. Curr Opin Chem Biol. 15:489-496.
- Edfeldt FN, Folmer RH, Breeze AL (2011). Fragment Screening to Predict Druggability (Ligandability) and Lead Discovery Success. Drug Discov. Today 16:284-7.
- Giordanetto F, Jin C, Willmore L, et al. (2019). Fragment Hits: What do They Look Like and How do They Bind? J. Med. Chem. 62:3381-3394.
- Erlanson D, Fesik S, Hubbard R. et al. (2016). Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov. 15:605-619.
- Lagoutte R, Patouret R, Winssinger N. (2017). Covalent inhibitors: an opportunity for rational target selectivity. Curr Opin Chem Biol. 39:54-63.
- Resnick E, Bradley A, Gan J, et al. (2019). Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 141:8951-8968.
- Ray S, Murkin AS. (2019). New Electrophiles and Strategies for Mechanism-Based and Targeted Covalent Inhibitor Design. Biochemistry. 58:5234-5244.
- Ostrem JM, Peters U, Sos ML, et al. (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 503:548-51.
- Johansson H, Tsai Y-CI, Fantom K, et al. (2019). Fragment-Based Covalent Ligand Screening Enables Rapid Discovery of Inhibitors for the RBR E3 Ubiquitin Ligase HOIP. J. Am. Chem. Soc. 141:2703-2712.
- Benns HJ, Wincott CJ, Tate EW, Child MA. (2021). Activity- and reactivity-based proteomics: Recent technological advances and applications in drug discovery. Curr. Opin. Chem. Biol. 60:20-29.
- Weerapana E, Wang C, Simon GM, et al. (2010). Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468:790-5.
- Backus K, Correia B, Lum K, et al. (2016). Proteomewide covalent ligand discovery in native biological systems. Nature 534:570–574.
- Hacker S, Backus K, Lazear M, et al. (2017). Global profiling of lysine reactivity and ligandability in the human proteome. Nature Chem. 9:1181–1190.
- Parker CG, Galmozzi A, Wang Y, et al. (2017). Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell. 168:527-541.
- Vinogradova EV, Zhang X, Remillard D, et al. (2020). An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell. 182:1009-1026.
- Kuljanin M, Mitchell DC, Schweppe, DK, et al. (2021). Reimagining high-throughput profiling of reactive cysteines for cell-based screening of large electrophile libraries. Nat. Biotechnol. 39:630–641.
- Zanon PRA, Yu F, Musacchio P, et al. (2021). Profiling the Proteome-Wide Selectivity of Diverse Electrophiles. ChemRxiv. Preprint. https://doi.org/10.26434/chemrxiv.14186561.v1
- Bar-Peled L, Kemper EK, Suciu RM, et al. (2017). Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell. 171:696-709.
Related topics
Assays, Drug Development, Drug Discovery, Molecular Targets, Protein, Proteomics, Screening, Small Molecules
Related organisations
UK Engineering and Physical Sciences Research Council