Dynamic model of SARS-CoV-2 S protein reveals vaccine targets
Posted: 7 April 2021 | Victoria Rees (Drug Target Review) | No comments yet
A model of the SARS-CoV-2 Spike protein surface has uncovered vulnerabilities that researchers say could inform the development of COVID-19 vaccines.
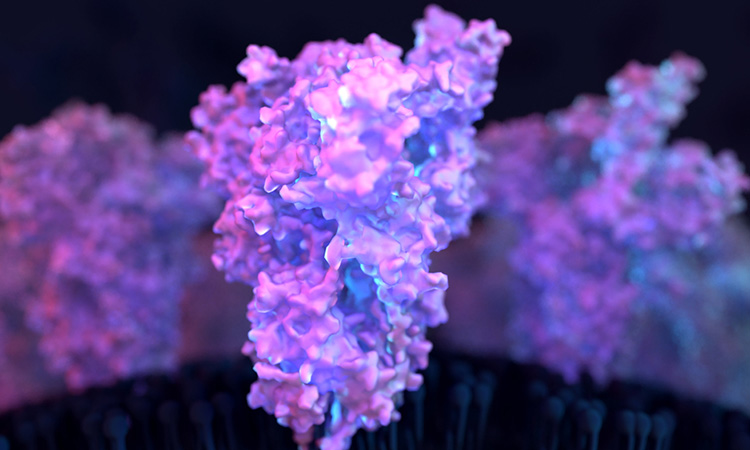
A new, detailed model of the surface of the SARS-CoV-2 Spike (S) protein has revealed previously unknown vulnerabilities that could inform the development of vaccines. The study was conducted at the Max Planck Institute of Biophysics, Germany.
According to the researchers, a key feature of SARS-CoV-2 is its S protein, which extends from its surface and enables it to target and infect human cells. Extensive research has resulted in detailed static models of the S protein, but these models do not capture the flexibility of the protein itself nor the movements of protective glycans – chains of sugar molecules – that coat it.
To support vaccine development, the team aimed to identify novel potential target sites on the surface of the S protein. To do so, they developed molecular dynamics simulations that capture the complete structure of the S protein and its motions in a realistic environment.
These simulations show that glycans on the S protein act as a dynamic shield that helps the virus evade the human immune system. Similar to car windshield wipers, the glycans cover nearly the entire S protein surface by flopping back and forth, even though their coverage is minimal at any given instant.
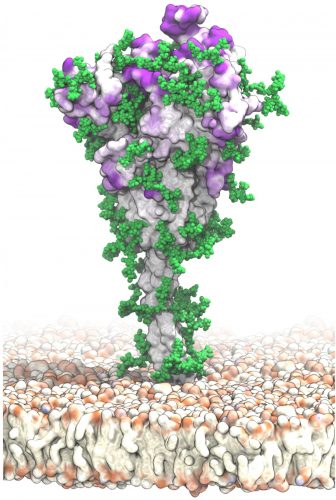
In this visualisation of antibody target sites, the SARS-CoV-2 S protein is tethered to the viral membrane with a slender stalk. Patches of intense purple colour at the surface of S indicate potential target sites for antibodies that are not protected by the glycans – chains of sugar molecules – shown in green. These binding sites and their accessibility were identified with molecular dynamics simulations that capture the complete structure of the spike protein and its motions in a realistic environment [credit: Mateusz Sikora, Sören von Bülow, Florian E. C. Blanc, Michael Gecht, Roberto Covino and Gerhard Hummer].
By combining the dynamic S protein simulations with bioinformatic analysis, the researchers identified spots on the surface of the proteins that are least protected by the glycan shields. Some of the detected sites have been identified in previous research, but some are novel. The vulnerability of many of these novel sites was confirmed by other research groups in subsequent lab experiments.
“We are in a phase of the pandemic driven by the emergence of new variants of SARS-CoV-2, with mutations concentrated in particular in the S protein,” said lead researcher Mateusz Sikora. “Our approach can support the design of vaccines and therapeutic antibodies, especially when established methods struggle.”
The researchers say that the method developed for this study could also be applied to identify potential vulnerabilities of other viral proteins.
The study was published in PLOS Computational Biology.
Related topics
Informatics, Molecular Targets, Protein, Proteomics, Structural Biology, Vaccine
Related conditions
Covid-19
Related organisations
Max Planck Institute of Biophysics
Related people
Mateusz Sikora