High-throughput screening as a method for discovering new drugs
Posted: 4 June 2020 | Mathew Lloyd (University of Bath) | No comments yet
High-throughput screening is a common method of identifying lead compounds for drug development. The most common targets are enzymes – catalytic proteins that perform chemical reactions in the cell. In this article, Matthew Lloyd discusses the opportunities and challenges associated with this approach.

High-throughput screening (HTS) is a commonly used method to identify bioactive compounds. The idea is that the function of a target (usually a protein) is measured, with a library of compounds screened for their ability to trigger and influence the desired change. Screening is often performed against an isolated protein using in vitro methods, although cellular systems are also quite commonly used. In academic settings, such campaigns use libraries containing thousands of compounds, while the pharmaceutical industry often leverages libraries with millions of compounds. The objective of HTS is to identify ‘lead’ compounds that can be developed into drugs, and occasionally useful drugs will be identified.1 A key advantage of using HTS is that compound discovery is driven by functional activity rather than known binding modes; this means that novel compounds are often more diverse when compared to compounds discovered through structure-based or rational design, in terms of their chemical structures and mechanisms of action. Consequently, HTS is often used to discover new bioactive compounds.2

The most common targets used in HTS campaigns are enzymes – catalytic proteins that promote chemical reactions within the cell.2 This is not only due to enzymes being well‑established drug targets for many diseases, but also because their catalytic reactions make them particularly amenable to drug discovery research using HTS. Kinases, which are responsible for phosphorylation of protein and non-protein substrates, are particularly common enzyme targets – accounting for 30 percent of all targets in an analysis of recent campaigns.2 This reflects the importance of kinases in many cellular processes and their significance in a large number of common diseases including cancer, inflammatory conditions, neurological diseases, dementia and depression.
A number of different assays can be used to measure enzymatic activity, but fluorescence-based methods, including fluorescence anisotropy and Förster Resonance Energy Transfer (FRET), are the most common due to their sensitivity, ease and adaptability to HTS formats.3 However, it is well known that the readout from fluorescent assays can be easily interfered with by fluorescent quenchers, coloured compounds and other mechanisms.3-5 In addition, many library compounds form aggregates onto which enzymes and/or substrates can adhere, resulting in false‑positive inhibition.6 This is the most common mechanism through which Pan-Assay Inference Compounds (PAINs) exert their effects,6 although such behaviour is heavily dependent on the assay conditions and it is notoriously difficult to predict whether this will occur in a particular assay. Consequently, it is common practice to run HTS assays in the presence of low concentrations of detergent such as Triton X-100 to prevent aggregate formation.7,8 Due to the high prevalence of false-positives in HTS, it is also common practice to rescreen all of the ‘hits’ identified in an initial screen by using another assay. Ideally this second assay will include a different substrate, inhibitor preparation and readout format as this minimises the risk of artefacts giving rise to false-positive results. The chemical structures of the ‘hits’ remaining after this confirmatory screen will be reconfirmed and evaluated for their novelty and tractability for development. It is common for less than 0.1 percent of the original library compounds in the initial HTS screen to make it this far, as most will be inactive or false positives. However, it is not uncommon to identify dozens, if not hundreds, of novel ‘lead’ compounds from an HTS, which can be potentially developed by systematic variation of structural features.
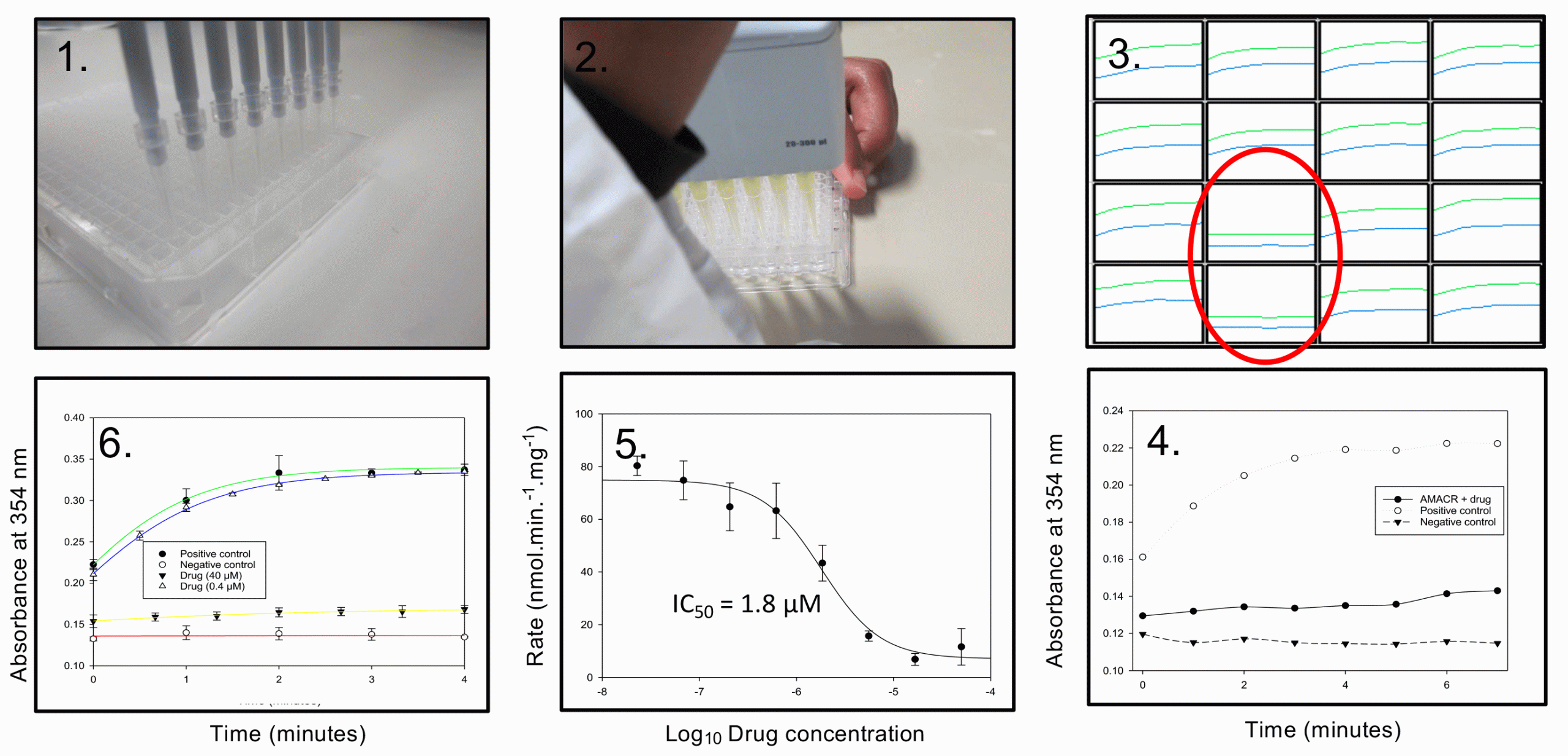
Figure 1: 1. Inhibitors are obtained as DMSO solutions in 384 plates; 2. Inhibitor solution is pipetted into a 96-well microtitre plate, followed by addition of enzyme and substrate; 3. Reaction is monitored with time, inhibitors are identified by a diminished increase in absorbance at 354nm (wells in red circle); 4. Comparison of inhibited reaction with positive (no inhibitor) and negative (no enzyme) controls; 5. Determining IC50 value using a dose-response curve; 6. Determining reversibility of inhibition using rapid dilution of drug, showing activity was restored upon ‘removing’ drug.
We recently reported an HTS campaign against α-methylacyl-CoA racemase (AMACR; P504S),9 a novel drug target for prostate cancer and several other cancers. AMACR catalyses a reversible epimerisation reaction and many of the published assay methods are not compatible with or are difficult to adapt to HTS formats.9 Consequently, only one previous study on AMACR using screening techniques has been published; this used a labour‑intensive radiochemical assay, which identified several non-specific proteininactivating agents as drugs.10 We developed a convenient colorimetric assay, based on the irreversible elimination of 2,4-dinitrophenolate from an unnatural substrate,11 which was adapted for use in HTS (Figure 1). For convenience, reactions were performed in 96-well plates (rather than the 384- or 1,535-well plates commonly used in industry) and colour development was monitored with time. Use of a continuous assay made it easier to interpret the results, as many library compounds absorbed at 354nm – the wavelength used to measure 2,4-dinitrophenolate formation. A substrate concentration close to its Km value was used (‘balanced conditions’12), allowing the identification of many different types of inhibitors. Using this method, we screened 20,387 different drug-like compounds.9
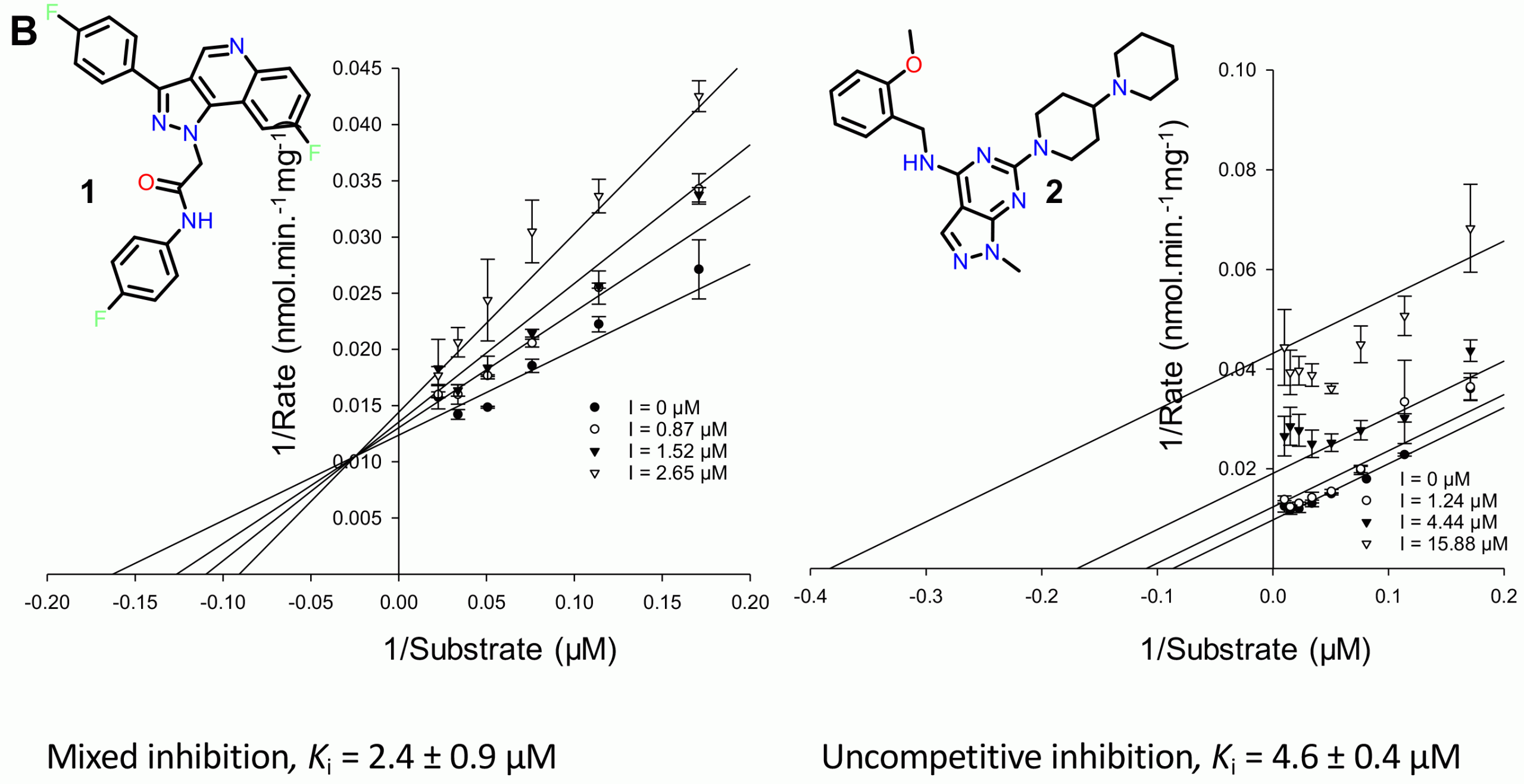
Figure 2: Determination of inhibition mode for the new inhibitors
As a result of this study,9 we identified two previously unknown families of AMACR inhibitors, the pyrazoloquinolines and pyrazolopyrimidines (Figure 2). Further characterisation revealed that these compounds were either mixed competitive or uncompetitive inhibitors (Figure 2), although their potency was highly dependent on the exact conditions used for the assay. Uncompetitive inhibition is rare because it arises when the inhibitor binds exclusively to the enzyme‑substrate complex, but offers significant advantages as inhibition is not lost due to substrate build-up within cells as a result of metabolic pathway blockages.9 This is not the case with competitive and mixed competitive inhibition (the two most commonly observed types), where inhibition can be overcome by increasing substrate concentrations within the cell.
However, further testing of these compounds in prostate cancer cells failed to show any reduction in cell growth and hence these drugs could not be easily developed into new treatments. Nevertheless, the study demonstrates the potential of HTS for identifying AMACR inhibitors as drug compounds.
About the author
Matthew Lloyd read Biological Chemistry at the University of Leicester followed by a DPhil (PhD) degree at the University of Oxford working on antibiotics until 1992. After several years of working as a post-doctoral researcher at Brown University, US and Oxford, Matthew joined the Department of Pharmacy and Pharmacology at the University of Bath in 2002. He is currently a Senior Lecturer (Associate Professor) in the Department. His research focuses on enzymes using techniques from chemistry, biochemistry and molecular biology to understand disease states and develop new treatments, especially for cancer.
References
1. Siegel D, Hui HC, Doerffler E, Clarke MO, Chun K, Zhang L, Neville S, Carra E, Lew W, Ross B, Wang Q, Wolfe L, Jordan R, Soloveva V, Knox J, Perry J, Perron M, Stray KM, Barauskas O, Feng JY, Xu YL, Lee G, Rheingold AL, Ray AS, Bannister R, Strickley R, Swaminathan S, Lee WA, Bavari S, Cihlar T, Lo MK, Warren TK, Mackman RL. J. Med. Chem., 2017, 60, 1648-1661. 2. Brown DG, Bostrom J. J. Med. Chem., 2018, 61, 9442-9468.
3. Gul S, Gribbon P. Expert Opin. Drug Discovery, 2010, 5, 681-690.
4. Beher D, Wu J, Cumine S, Kim KW, Lu SC, Atangan L, Wang M. Chem. Biol. Drug Des., 2009, 74, 619-624.
5. Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, Qiu XY, Stockman B, Thanabal V, Varghese A, Ward J, Withka J, Ahn K. J. Biol. Chem., 2010, 285, 8340-8351.
6. Baell JB, Holloway GA. J. Med. Chem., 2010, 53, 2719-2740.
7. Jadhav A, Ferreira RS, Klumpp C, Mott BT, Austin CP, Inglese J, Thomas CJ, Maloney DJ, Shoichet BK, Simeonov A. J. Med. Chem., 2010, 53, 37-51.
8. Feng BY, Simeonov A, Jadhav A, Babaoglu K, Inglese J, Shoichet BK, Austin CP. J. Med. Chem., 2007, 50, 2385-2390.
9. Petrova YD, Wadda K, Nathubhai A, Yevglevskis M, Mitchell PJ, James TD, Threadgill MD, Woodman TJ, Lloyd MD. Bioorg. Chem., 2019, 92, article 103264.
10. Wilson BAP, Wang H, Nacev BA, Mease RC, Liu JO, Pomper MG, Isaacs WB. Mol. Cancer Ther., 2011, 10, 825-838.
11. Yevglevskis M, Lee GL, Nathubhai A, Petrova TD, James TD, Threadgill MD, Woodman TJ, Lloyd MD. Chem. Commun., 2017, 53, 5087-5090.
12. Copeland RA. Anal. Biochem., 2003, 320, 1-12.
Related topics
Analysis, Analytical Techniques, Disease Research, Drug Development, Drug Targets, Enzymes, High-Throughput Screening (HTS), Lead Generation
Related conditions
Cancer, Prostate cancer
Related organisations
ForteBio, Horizon Discovery, Merck, Ncardia