Deep learning network used to model protein complexes
Posted: 13 January 2020 | Victoria Rees (Drug Target Review) | No comments yet
A new study has developed a deep learning approach that analyses protein interactions, which could improve the design of drugs in the future.
Researchers have designed a novel approach to use deep learning to improve their understanding of how proteins interact in the body. According to the team, their findings could result in more accurate structure models of protein interactions in various diseases and better drug designs that target protein interactions.
…this information can be used in the creation of targeted drugs to block certain protein-protein interactions”
The study, conducted at Purdue University, US, designed a system called the docking decoy selection with voxel-based deep neural network (DOVE). This applies deep learning principles to virtual models of protein interactions.
“To understand molecular mechanisms of functions of protein complexes, biologists have been using experimental methods such as X-rays and microscopes, but they are time- and resource-intensive efforts,” said Daisuke Kihara, a professor of biological sciences and computer science in Purdue’s College of Science, who leads the research team. “Bioinformatics researchers in our lab and other institutions have been developing computational methods for modelling protein complexes. One big challenge is that a computational method usually generates thousands of models and choosing the correct one or ranking the models can be difficult.”
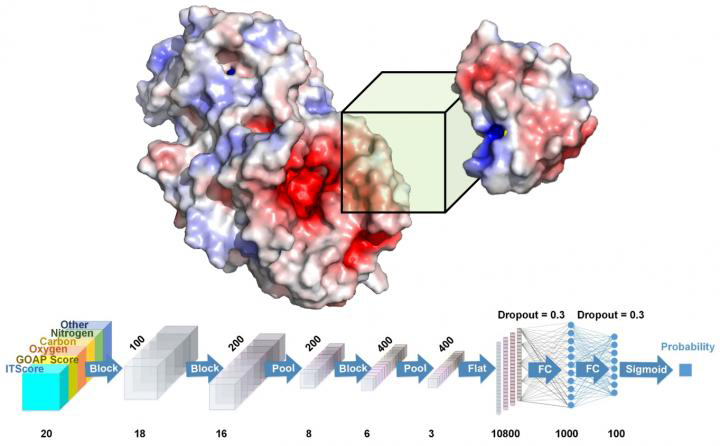
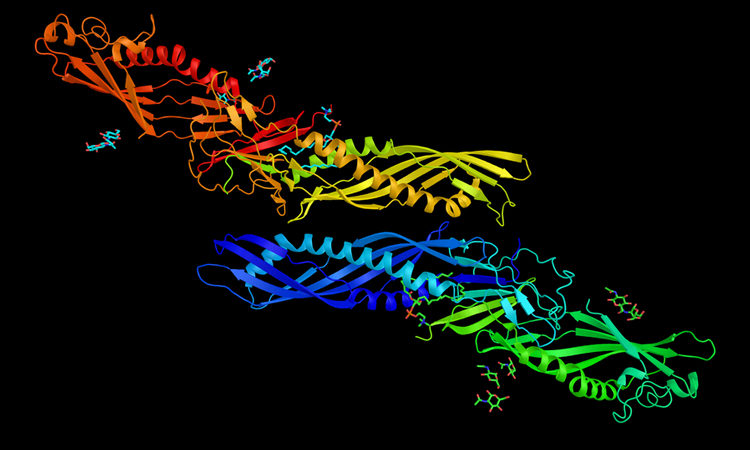
DOVE, created by Purdue researchers, captures structural and energetic features of the interface of a protein docking model with a 3D box and judges if the model is more likely to be correct or incorrect using 3D convolutional neural network (credit: Daisuke Kihara/Purdue University).
The researchers used DOVE to scan the protein-protein interface of a model and then distinguish and capture structural features of correct and incorrect models.
“Our work represents a major advancement in the field of bioinformatics,” said Xiao Wang, a graduate student and member of the research team. “This may be the first time that researchers have successfully used deep learning and three dimensional (3D) features to quickly understand the effectiveness of certain protein models. Then, this information can be used in the creation of targeted drugs to block certain protein-protein interactions.”
The results were published in Bioinformatics.
Related topics
Bioinformatics, Imaging, Informatics, Protein, Proteogenomics, Proteomics
Related organisations
Purdue University
Related people
Professor Daisuke Kihara, Xiao Wang