Revealed protein structure could aid in Huntington’s disease treatment development
Posted: 15 February 2021 | Victoria Rees (Drug Target Review) | No comments yet
Using NMR spectroscopy, researchers have partially observed the structure of heat shock proteins that bind to proteins that cause Huntington’s disease.
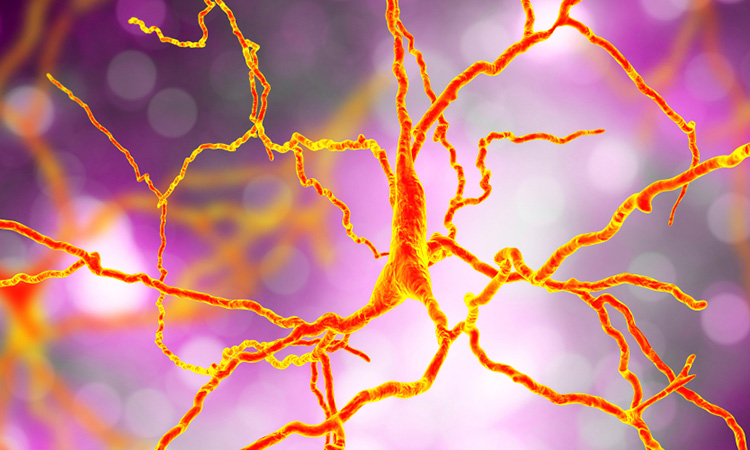
New research from the University of Groningen, the Netherlands, and the University of Texas, US, has partially resolved the structure of heat shock proteins (Hsps) that bind to the aggregating proteins that cause Huntington’s disease, providing insight into how they work.
According to the researchers, Hsps are produced by cells that are exposed to stressful conditions. The Hsp family is diverse and quite a few of the proteins function as chaperones; they help other proteins to fold (or re-fold after being damaged) in the correct way.
“These proteins can assist in folding thousands of different proteins. To this end, they use co-chaperones with specific binding abilities,” explained lead researcher Associate Professor Patrick van der Wel, at the University of Groningen.
One class of Hsps, Hsp40, helps to suppress protein aggregates like those that appear in Huntington’s disease. Some of these Hsp40 proteins bind specifically to aggregating proteins with a lot of repeated glutamine amino acids, like the faulty protein found in Huntington’s disease. One of these Hsp40 proteins is called DnaJB8 and this was the protein studied by Van der Wel and his colleagues.
“In order to understand the action of DnaJB8, we need to know what it looks like,” said Van der Wel. However, it is difficult to resolve the structure of this type of protein. “It appears as a dimer or an oligomer, so a number of these protein units work together, but their structure is not really ordered.”
As Van der Wel is specialised in solid-state NMR spectroscopy, he and the team investigated the NMR signals of two connected carbon atoms in DnaJB8 to observe how they interact with other atoms in the molecule. The measured spectrum of the carbon atoms could therefore show in which amino acid they are located.
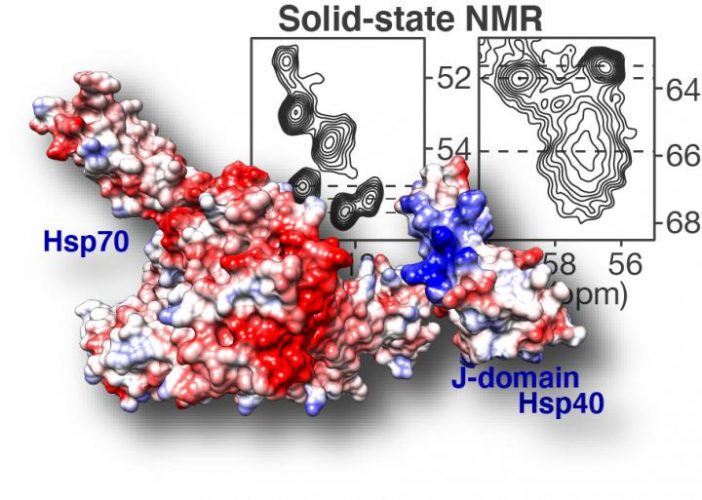
Solid-state NMR spectroscopy (top right) highlights how DnaJB8 chaperone protein blocks itself. In the displayed NMR data each signal comes from an amino acid in a key part of the chaperone protein: its J-domain. Based on the shape and steepness of the contour lines, we know that this domain is trapped by the chaperone itself, keeping it from doing its job. Fortunately it is also shown how this inactivated state can be disrupted, revealing an intricate orchestration in how chaperone proteins work together to fight and prevent diseases such as Huntington’s disease [credit: Patrick van der Wel, University of Groningen].
Through a series of experiments, Van der Wel was able to determine which domains are inside the DnaJB8 protein and which are available on the outside. The experiments suggested that the so-called ‘J domain’ of DnaJB8 was able to switch between being stuck and being accessible. According to the team, this is important because this part of the DnaJB8 protein is responsible for turning on the Hsp70 protein, which can prevent the protein aggregates from forming. They conclude that there seems to be a ‘switch’ in DnaJB8 that controls this interaction with Hsp70. This switch was found to be located in a domain of DnaJB8 whose exact role was previously unclear.
“Thus, our hypothesis based on the structure was that the DnaJB8 is inactive until it binds to the faulty proteins and that it then attracts Hsp70,” said Van der Wel. A series of simulations and experiments at the University of Texas confirmed this idea and yielded a detailed model of how these proteins work together.
As DnaJB8 is mainly found in the testes, a very similar protein called DnaJB6 that is present in the brain, could be used to protect against glutamine-rich proteins that aggregate in the brain cells of patients and cause Huntington’s disease.
“It may take many more years, but now that we understand how this process works, it could help us to find a way to enhance the activity of DnaJB6, which could reduce the protein aggregates that cause the disease,” said Van der Wel.
The results were published in Nature Communications.
Related topics
Drug Targets, Heat Shock Proteins (HSPs), Nuclear Magnetic Resonance (NMR), Protein, Proteomics, Spectroscopy, Targets
Related conditions
Huntington's disease
Related organisations
Texas University, University of Groningen
Related people
Associate Professor Patrick van der Wel