Targeting SARM1 to protect vision across retinal degenerative diseases
Posted: 23 June 2020 | Dr Ema Ozaki (Trinity College Dublin), Dr Sarah Doyle (Trinity College Dublin) | No comments yet
Dr Sarah Doyle and Dr Ema Ozaki outline their research into SARM1 and why it presents an attractive target for treating retinal degeneration.
Retinal degenerative diseases, including age-related macular degeneration, retinitis pigmentosa and diabetic retinopathy are the leading causes of severe visual impairment and eventual blindness, affecting over 200 million people worldwide. There are a wide range of lifestyle factors that can promote retinal degeneration such as age, diet and smoking, in addition to over 200 genes that are implicated in inherited retinal diseases; however, in cases end with the ultimate damage, malfunction and death of light sensitive cells in the retina called photoreceptors. Photoreceptors, comprising of rod and cone cells, initiate the visual phototransduction process by absorbing photons of light and converting them into electric impulses that are sent to the visual cortex in the brain to create an image. Currently, there are very limited treatment options for many of these blinding conditions; therefore, identifying pro-degenerative or pro-survival mechanisms in these photoreceptor cells is key for discovering new therapeutic targets. This article explores the potential that the executioner molecule SARM1 presents as one such novel therapeutic target.1
Background of SARM1
Sterile alpha and Toll/interleukin-1 receptor motif–containing 1 (SARM1) was originally identified as a member of the Toll/IL-1 Receptor (TIR) domain-containing superfamily that can regulate innate immune activation.2 However, SARM1 has now come into focus in studies of the central nervous system (CNS) and brain where it has a critical function in removing damaged nerve axons and triggering neuronal cell death.3-5
 Endogenous SARM1 promotes neuronal cell death in response to a wide range of insults, including oxidative stressors, mitochondrial poisons, oxygen-glucose deprivation, injury and neurotrophic viruses, many of which have been implicated in retinal degenerative disease processes.6-7 Mechanistically, SARM1-mediated cell death appears to be different to other forms of cell death including apoptosis, necrosis and necroptosis. The TIR domain of SARM1 possesses intrinsic enzymatic activity and its activation leads to the cleavage and precipitous loss of the essential metabolite NAD+, resulting in a metabolic crisis and triggering neuronal cell degeneration.8-11
Endogenous SARM1 promotes neuronal cell death in response to a wide range of insults, including oxidative stressors, mitochondrial poisons, oxygen-glucose deprivation, injury and neurotrophic viruses, many of which have been implicated in retinal degenerative disease processes.6-7 Mechanistically, SARM1-mediated cell death appears to be different to other forms of cell death including apoptosis, necrosis and necroptosis. The TIR domain of SARM1 possesses intrinsic enzymatic activity and its activation leads to the cleavage and precipitous loss of the essential metabolite NAD+, resulting in a metabolic crisis and triggering neuronal cell degeneration.8-11
SARM1 can delay rod and cone photoreceptor cell death
…[an] AAV targeting SARM1 could be effective in retinal disease models”
Data from the Human Proteome Map,12 a mass spectrometry-based proteomics resource, reveals that apart from the foetal brain, SARM1 is most highly expressed in the adult retina when compared to all other human tissues. Moreover, its high abundancy in photoreceptor cells specifically was demonstrated when SARM1 expression levels in the retina of wild-type mice were compared with levels in mice lacking photoreceptor cells, aged rhodopsin knockout (rho-/-) mice; the latter had significantly less SARM1 retinal expression – indicating that SARM1 is abundantly expressed in the photoreceptors. To examine whether SARM1 plays a role in regulating cell death in these cells, an active SARM1 construct was overexpressed in the 661W photoreceptor-like cell line. Indeed, overexpression of enzymatically active SARM1 resulted in the death of these cells and using retinal tissue explants, SARM1 activation was shown to coincide with the destruction of NAD+ pools in the photoreceptors.
The role of SARM1 was examined in the rho-/- in vivo mouse model of retinal degeneration. This model is useful for studying the effects of manipulating aspects of cell death/cell survival processes because young rho-/- mice of postnatal day 21 have comparative numbers of photoreceptor nuclei and photoreceptor layer thickness to wild-type mice. However, rho-/- mice fail to build rod photoreceptor outer segments and by ~12 weeks of age all photoreceptor cells are lost. Cone visual function measured by electroretinogram (ERG), is relatively normal until seven weeks of age, indicating that the early loss in photoreceptors are solely in rods.13 The absence of cone ERGs by 12 weeks of age indicates that early rod degeneration is followed by a rapid degeneration in cones between eight and 12 weeks; these characteristics make it an ideal model to examine both rod and cone photoreceptor cell deaths individually.
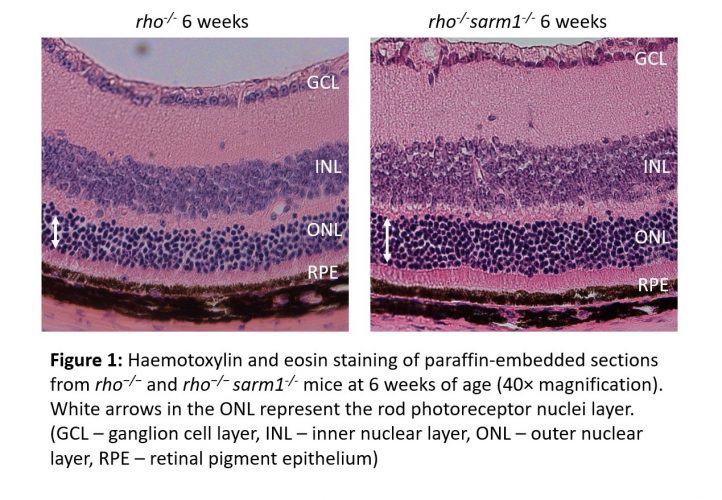
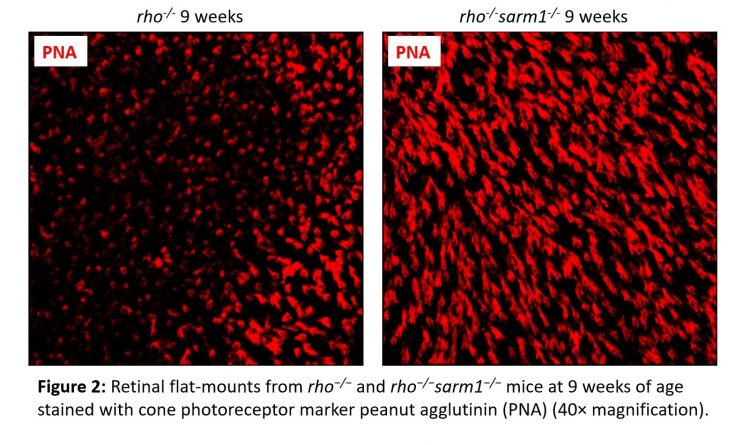
The generation of rho−/−sarm1−/− double knockout mice demonstrated that SARM1 deficiency was protective against rod photoreceptor cell death, with a higher number of rod photoreceptor cell nuclei present from six weeks of age (Figure 1). This was further supported by delayed retinal thinning measured by optical coherence tomography. Furthermore, ERGs revealed that the loss of SARM1 preserved cone visual function in the surviving photoreceptors from nine to 12 weeks of age and a greater number of cone cells were observed histologically (Figure 2). A decreased NAD+ pool was observed in retinal explants from rho-/- mice compared to rho−/−sarm1−/- double knockout mice, indicating that activation of SARM1 in the rho-/- mice results in the loss of essential metabolic co-factor NAD+.
Overall, the study provides compelling evidence that loss of SARM1 activity can promote both rod and cone cell survival and changes in the NAD+ pool implicate SARM1-mediated NAD+ depletion in the mechanism of cell death. In support of this, a recent study has highlighted the importance of NAD+ for vision in mice as photoreceptor specific deletion of nicotinamide phosphoribosyltransferase (Nampt), the rate limiting enzyme in the major NAD+ biosynthetic pathway, caused retinal degeneration and retinal NAD+ deficiency was observed as an early feature in multiple retinal degeneration mouse models.14
SARM1 as a drug target
This new research is particularly exciting for the development of future therapeutic development in the field of retinal degeneration. A recent study by Geisler et al. has developed a gene therapy approach for inhibiting SARM1 in vivo in a model of nerve transection.15 Geisler et al. use an adeno-associated virus (AAV) to deliver SARM1 constructs with point mutations that can act in a dominant negative fashion to inhibit the function of wild-type SARM1. Gene therapy is well known to be suited for treatment of retinal disease; the first gene therapy to receive US Food and Drug Administration (FDA) approval in 2017 was Luxturna, for patients with RPE65-mediated retinal dystrophy such as Leber congenital amaurosis and retinitis pigmentosa. Therefore it is a realistic expectation that a similar AAV targeting SARM1 could also be effective in retinal disease models.
…loss of SARM1 activity can promote both rod and cone cell survival”
In addition to SARM1 inhibition by gene therapy, recent pre-clinical data from Disarm Therapeutics have demonstrated that orally administered small molecule inhibitors of SARM1 enzymatic activity can prevent axonal cell death and preserve axonal structure and function in mouse model of chemotherapy-induced peripheral neuropathy.16 Finally, while mendelian inherited retinopathies are ideally suited to gene therapy, they are genetically heterogeneous with over 260 disease genes identified to date. The cost associated with design and generation of individualised AAV constructs for each of these is significant. SARM1 is activated by a broad range of injuries observed in retinal degeneration. Therefore, targeting a unifying pro-degenerative molecule such as SARM1 might present a cost-effective therapeutic avenue for protecting vision across multiple retinal degenerative diseases in the future.
About the authors
 Dr Ema Ozaki obtained her PhD in 2015 from the Department of Genetics in Trinity College Dublin and has since being working as a postdoctoral research fellow in the Department of Clinical Medicine in Trinity College Dublin.
Dr Ema Ozaki obtained her PhD in 2015 from the Department of Genetics in Trinity College Dublin and has since being working as a postdoctoral research fellow in the Department of Clinical Medicine in Trinity College Dublin.
 Dr Sarah Doyle is an Irish Research Council Laureate and Assistant Professor in Immunology in Trinity College Dublin.
Dr Sarah Doyle is an Irish Research Council Laureate and Assistant Professor in Immunology in Trinity College Dublin.
References
- Ozaki E, Gibbons L, Neto NG, Kenna P, Carty M, Humphries M, et al. SARM1 deficiency promotes rod and cone photoreceptor cell survival in a model of retinal degeneration. Life Sci Alliance. 2020;3(5).
- Carty M, Kearney J, Shanahan KA, Hams E, Sugisawa R, Connolly D, et al. Cell Survival and Cytokine Release after Inflammasome Activation Is Regulated by the Toll-IL-1R Protein SARM. Immunity. 2019;50(6):1412-24.e6.
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337(6093):481-4.
- Geisler S, Doan RA, Strickland A, Huang X, Milbrandt J, DiAntonio A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain. 2016;139(Pt 12):3092-108.
- Turkiew E, Falconer D, Reed N, Höke A. Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J Peripher Nerv Syst. 2017;22(3):162-71.
- Mukherjee P, Woods TA, Moore RA, Peterson KE. Activation of the innate signaling molecule MAVS by bunyavirus infection upregulates the adaptor protein SARM1, leading to neuronal death. Immunity. 2013;38(4):705-16.
- Summers DW, DiAntonio A, Milbrandt J. Mitochondrial dysfunction induces Sarm1-dependent cell death in sensory neurons. J Neurosci. 2014;34(28):9338-50.
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD⁺ destruction. Science. 2015;348(6233):453-7.
- Sasaki Y, Nakagawa T, Mao X, DiAntonio A, Milbrandt J. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD(+) depletion. Elife. 2016;5.
- Summers DW, Gibson DA, DiAntonio A, Milbrandt J. SARM1-specific motifs in the TIR domain enable NAD+ loss and regulate injury-induced SARM1 activation. Proc Natl Acad Sci U S A. 2016;113(41):E6271-e80.
- Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD(+) Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron. 2017;93(6):1334-43.e5.
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, et al. A draft map of the human proteome. Nature. 2014;509(7502):575-81.
- Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, et al. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15(2):216-9.
- Lin JB, Kubota S, Ban N, Yoshida M, Santeford A, Sene A, et al. NAMPT-Mediated NAD(+) Biosynthesis Is Essential for Vision In Mice. Cell Rep. 2016;17(1):69-85.
- Geisler S, Huang SX, Strickland A, Doan RA, Summers DW, Mao X, et al. Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J Exp Med. 2019;216(2):294-303.
- Krauss R, Bosnac T, Engber T, Devraj R, Hughes R. Small Molecule Inhibitors of SARM1 Prevent Axonal Degeneration in vitro and in vivo. Society for Neuroscience Annual Meeting; Chicago 2019.
Related topics
Drug Targets, Enzymes, Protein, Proteomics, Research & Development, Targets
Related conditions
diabetic retinopathy, Leber congenital amaurosis, Macular degeneration, retinal degeneration, Retinitis pigmentosa
Related organisations
Disarm Therapeutics, US Food and Drug Administration (FDA)