Exploring the intra-protein non-covalent interactions in the S1 domain of the SARS-CoV-2 S1 Spike protein
Posted: 11 September 2020 | Oscar Bastidas PhD (University of Minnesota) | No comments yet
The Spike (S) protein is a promising drug target in the quest to develop vaccines and antivirals for SARS-CoV-2 to treat and prevent COVID-19. Intra-protein interaction energy landscape mapping efforts have recently identified key interactions that may be crucial for the S protein’s ability to bind to receptor proteins on host epithelial cells – a precursor necessary for cellular infection. Consequently, characterising the interaction profile of the S protein might be key to disrupting these interactions with specifically engineered inhibitors. In this article, Oscar Bastidas details his latest research work, which follows an approach that could potentially lead to effective treatments and novel vaccine candidates in the fight against COVID-19.
IN AN EFFORT to identify specific inter- and intra-protein interactions that might serve as viable drug targets for the development and design of therapeutic agents for treating COVID-19, I have been conducting research on the Spike (S) protein of the SARS-CoV-2 virus. This involved using computational methods that probe the specific atom-atom interactions inside specified regions of this protein. For this study, the energetic mapping involved the input of a crystal structure of the protein (PDB accession: 6vsb) to analyse various regions within the chain. This resulted in the identification of atom-atom interactions strong enough to maintain structural elements and cohesion within the S protein for the specified region.
The methodology specifically looked at partial charge and Van der Waals interactions in the S1 and S2 domains of the trimeric S protein of SARS-CoV-2. The three main observations were: 1) there were significant energetic differences between the up-down and down-down states of the S1 domains of neighbouring protomers (inter-chain mapping) in the trimer, specifically, between the receptor binding domain (RBD) and the N-terminal domains (NTD); 2) the intra-chain dominant energy mappings within each protomer identified a significant “glue” point or possible “latch” for the down-state protomer between the S1 subdomain (SD1) and the RBD domain of the same protomer that was found to be completely missing in the up-state configuration; and 3) a three-residue sequence (ALA520-PRO521-ALA522) appeared in the mapping results between the up-down RBD-NTD inter-chain interactions that were associated with a turn structure in the RBD of the up-state protomer as well as appearing in the aforementioned latch identified from the intra-chain analysis.
Following my recently published study on Aβ42 amyloid fibrils,1 I analysed the recently published trimeric structure of SARS-CoV-2 (PDB ID: 6vsb) according to the Coulombic (charge and partial atomic charge) and Lennard-Jones (Born and Van der Waals forces) atom-atom interaction forces as laid out in the open-source energy mapping algorithm.2 This mapping algorithm efficiently parses the strongest non-covalent atom-atom interactions and their inter-atomic distances from structure file data, according to empirically established criteria based on the AMBER03 force field model, to ensure that all dominant interactions are accounted for. As in the aforementioned Aβ42 studies, the parsing criteria were taken as the upper limit of −0.1kT units for Lennard-Jones (Van der Waals) criteria and −0.3kT units for Coulombic interactions, although lower values can also be specified in the analysis of the mappings to further refine the results.2 Of the three chains the structure consisted of (chains A, B and C, where A was in the up state and B and C were in the down state), the inter-chain interaction mappings specifically looked at interactions between chains A and B for the up-down scenario and between chains B-C for the down-down interactions. For the intra-chain interactions, I also investigated the S1 and S2 domain interactions and the subdomains of S1 that included the RBD domain.
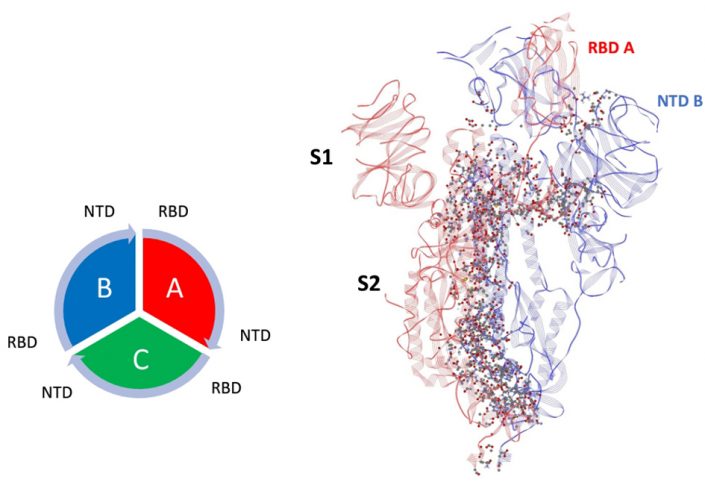
Figure 1: Dominant energy landscape for the interaction of chain A (Up; red) with chain B (Down; blue). The dominant atom-atom interactions are shown by ball and sticks. Also shown is the overall chain interaction configuration looking at the trimer from the top view.
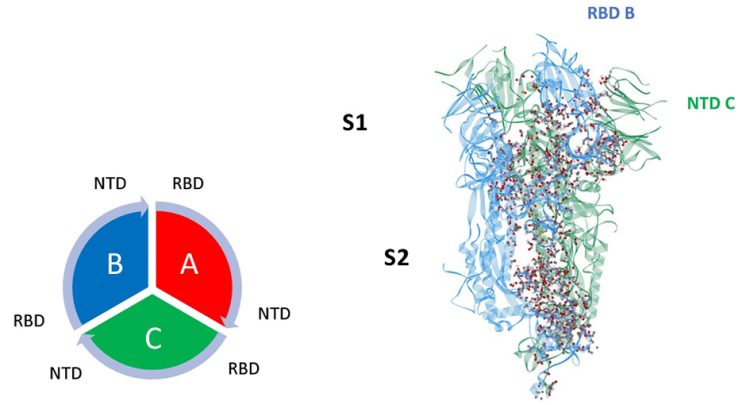
Figure 2: Dominant energy landscape for the interaction of chain B (Down; blue) with chain C (Down; green). The dominant atom-atom interactions are shown by ball and sticks.
Mappings of the dominant atom-atom interactions among the residues for the up-down (A-B) and down-down (B-C) states are illustrated in Figures 1 and 2, respectively. The majority of the dominant interactions were found to be within the S2 domain and the three-chain structure appears to be greatly stabilised by this feature. There were several stabilising interactions involving the SD1/SD2 domain of any given protomer with the S2 and NTD domain of neighbouring protomers. The pattern of the S1-S1 inter-chain interactions involving the RBD of any chain to the NTD of its adjacent neighbour is potentially important, as shown in Figures 1 and 2. The inter-chain interactions between the RBD with the NTD of its neighbour protomer for both the up-down (A-B) and down-down (B-C) interactions, shown in Figures 3 and 4, respectively, were examined. Somewhat surprisingly, the up-down interactions were stronger and more numerous overall than the down-down interactions of these close neighbour domains. Additionally, the energetically dominant up-down RBD-NTD interactions involved three additional residues (ALA520-PRO521-ALA522) not seen in the down-down cross-chain interactions. These three residues, however, will be shown to play a stabilising intra-chain role for the down state protomer.
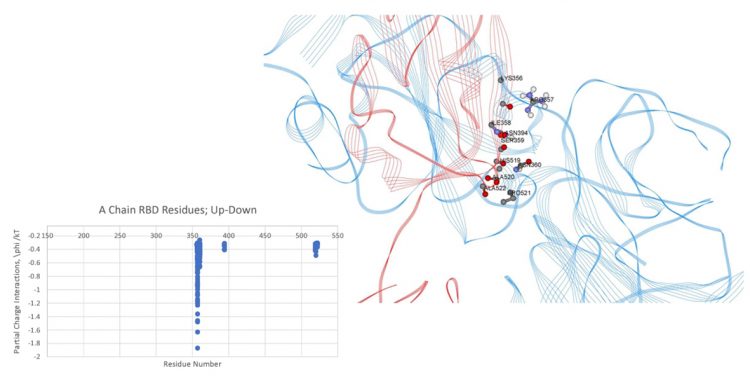
Figure 3: Dominant energy landscape for the interaction of the RBD chain A (Up;red) with NTD chain B (Down;blue); oblique, top view. Only A chain RBD residue/atoms are shown.
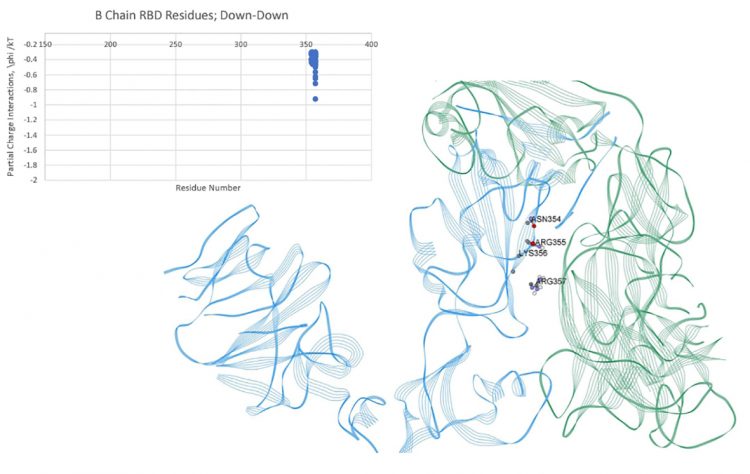
Figure 4: Dominant energy landscape for the interaction of the RBD of chain B (Down;blue) with the NTD of chain C (Down;green); oblique, top view. Only B chain residue/atoms are shown.
The intra-chain interaction mappings next sought to identify those interactions that were different between the up state versus the down state within a protomer subunit. Looking at the mapping of the S1 domain to the S2 domain, no discernable differences in the energetic interactions between the up and down states were observed, which may not be too surprising considering that the RBD of S1 (residues 319-527) is distal to the S2 domain in either the up or down conformation. Additionally, the proximal S1 domain beyond the RBD, SD1/SD2 domains (residues 533-677), also showed no discernable differences between the up and down states in its interaction with S2. Overall, the energetic mappings between the up and down states for the two S1 domains, RBD-SD1/SD2, appeared very similar, with just one important difference. For the down state protomer, conspicuous, strong energetic interactions between the R group of GLN564 in the SD1 domain with backbone atoms of the three sequential residues ALA520-PRO521-ALA522 in the RBD domain were observed, which were missing in the up state mappings due to the more distal state of the RBD in that conformation. These specific atom-atom interactions are shown in more detail in Figures 5 and 6. It is worth noting that the PRO 521 interactions are dominated by Van der Waals interactions in both instances. Interestingly, these are exactly the same three residues of the up-RBD that participate in the set of dominant energetic interactions with the down-NTD of its neighbour protomer along the up-down interface (Figure 3). Thus, these three dominant energy residues appear to play different roles depending on the up or down state of the protomer, which was somewhat surprising.
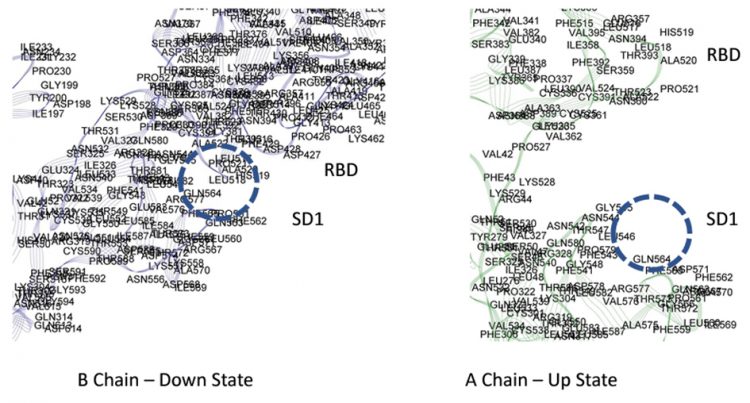
Figure 5: Dominant energy landscape for the partial charge interactions between SD1 and RBD of of chains A and B showing a possible latch for the B chain.
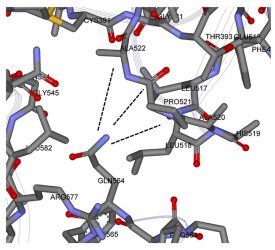
Figure 6: Zoomed view of possible molecular latch between the RBD and SD1 domain: ALA520-PRO521-ALA522 with GLN 564.
In order to determine if these strong, but few residue interactions could possibly function as a molecular latch for the down state protomer, I conducted some preliminary all-atom molecular dynamics simulations using powerful supercomputers at NASA’s Ames Research Center in California’s Silicon Valley with my collaborator, Michael Peters, from Virginia Commonwealth University’s College of Engineering. The simulations covered a total period of 0.1 µsecond for a wildtype single S1 chain and a single point mutant S1 chain, both in the same initial down protomer state from PDB ID 6vsb. For the mutant (single-point mutation), the R group glue point atoms of GLN 564 were eliminated by replacing that amino acid with GLY 564. The three-residue sequence ALA520-PRO521-ALA522 in the RBD domain or any other residues throughout the S1 chain were not altered. Since the specific interest here was investigating structural change, alanine screening was not used, which is often employed instead for biological activity mutation analysis while preserving secondary structure.
Trajectory analysis revealed the early release of the RBD from SD1 (within a few nanoseconds) and overall hinge opening and distal release of the RBD for this single point mutant, whereas the hinge angle and RBD relative positions were preserved in the wild state (Figure 7). For completeness, RMSF values for both wild and mutant were also determined and indicated a relatively high degree of flexibility for the mutant S1 domain (RMSF values ranged from five to 25˚A). These dynamic results are preliminary and additional simulations of the complete trimer are presently ongoing. Nevertheless, as the static energy mapping results indicate, inter-chain interactions in the trimer may be potentially stabilised by crosschain interactions and these could play a critical role in the overall conformation of the trimer and the up versus down protomer states.
Figure 7: Initial dynamic test of possible latch over a period of approximatley 0.1µ sec. Blue – Wild; Red-Mutant.
In all, these studies provide insight into the characterisation of the cohesive inter-atomic forces at play in the stabilisation and dynamics of the sub-units of the SARS-CoV-2 S protein. Given the role this protein plays in the initial stages of cellular infection, it is an important drug target for the development of treatments for COVID-19. From these studies alone, we postulate that a class of peptide-based viral-cell binding inhibitors can be immediately engineered to selectively target the aforementioned regions given their conformational properties. Besides probing the dynamic characteristics of the complete trimer, we also note that future work with this system will involve analysing the dynamic behaviour of the S protein during the binding process between the virus and host cell. With such a complete characterisation campaign carried out, therapeutic agents could therefore be better designed to out-perform therapeutic species identified by brute-force drug re-purposing screening efforts.
The study can be found at: www.ncbi.nlm.nih. gov/pmc/articles/PMC7263490/
About the author
Oscar Bastidas is a postdoctoral researcher at the University of Minnesota working in the field of drug design and pharmaceutical research and development. He uses both experimental and computational methods to investigate the intricacies of proteinprotein interactions and their dynamic behaviour. His specific research interests include using peptides as drugs in the application of viral diseases and employing machine learning and big data analytics in drug design.
References
- Bastidas OH, Green B, Sprague M, Peters MH. Few Ramachandran Angle Changes Provide Interaction Strength Increase in Aβ42 versus Aβ40 Amyloid Fibrils. Scientific reports. 2016 Nov 3;6(1):1-0.
- Krall A, Brunn J, Kankanala S, Peters MH. A simple contact mapping algorithm for identifying potential peptide mimetics in protein–protein interaction partners. Proteins: Structure, Function, and Bioinformatics. 2014 Sep;82(9):2253-62.
Related topics
Drug Repurposing, Drug Targets, Peptide Therapeutics, Protein, Proteomics, Research & Development, Targets
Related conditions
Covid-19
Related organisations
Fluidic Analytics, NASA, NASA Ames Research Center, Virginia Commonwealth University
Related people
Michael Peters