Tweaking synonymous sites for gene therapy and vaccines
Posted: 30 November 2020 | Professor Laurence D Hurst (University of Bath) | No comments yet
Professor Laurence D Hurst explains why understanding the nucleotide mutations in viruses, including SARS-CoV-2, can have significant implications for vaccine design.
With 61 codons specifying 20 amino acids, some can be encoded by more than one codon and it is often presumed that it does not matter which one a gene uses. When I first studied genetics, some books I read taught that mutations between such alternative codons (eg, GGA->GGC, both giving glycine) were called “synonymous” mutations, while others referred to them as “silent” mutations. However, are synonymous mutations really silent – meaning they are identical in terms of fitness and function? Although they may specify the same amino acid, does that mean they are all the same?
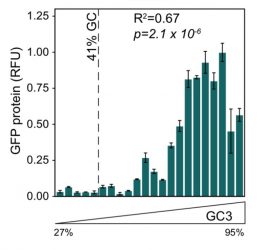
Figure 1: Intronless GFP transgene expression is higher for variants of GFP with higher GC content at synonymous sites5
Perhaps one of the biggest surprises over recent years has been the discovery that versions of the same gene, differing only at synonymous sites, can not only have different properties, but effects that are not modest.1-5 For example, two versions of green fluorescent protein (GFP) differing only at synonymous sites can have orders of magnitude differences in their expression level.4 We similarly recently discovered that for an intronless transgene to express in human cell lines it needs to be GC rich, which can be achieved by altering the synonymous sites,5 as seen in Figure 1. It is no accident, we suggest, that the well-expressed endogenous intronless genes in humans (such as histones) are all GC rich and that our functional retrogenes tend to be richer in GC content than their parental genes.
The realisation that synonymous sites matter has clear relevance to the design of transgenes or other artificial genes, be these for experiments, gene therapy, protein production (eg, in bacteria) or for vaccine design. In the case of vaccines, we might wish to modulate a viral protein to be effectively expressed in human cells to illicit a strong and robust immune response.6 Conversely to the design of attenuated vaccines, we seek to produce a tuned down version of the virus that can function but is weak.7
The challenge is knowing not just which synonymous sites can be altered but knowing how they should be altered. One approach is mass randomisation – try many alternatives and see what works.4,8,9 In principle this is fine, but this approach requires many randomisations, which is still technically difficult for long attenuated viruses. An alternative strategy that we have been exploring is to let nature tell us; we can apply tools and ideas from population genetics to better understand what natural selection favours and disfavours and in turn to estimate the strength of selection.
…it will be interesting to see if we can learn a lesson from nature as to how to weaken a virus”
Estimation of the strength of selection is possible from knowledge of the site frequency spectrum, (ie, how common variants are) from which we can infer the distribution of fitness effects (DFE). If a site is under strong purifying selection, then mutations may occur in the population but these are rapidly eliminated, so variants are always rare. By contrast, if they are selectively neutral, we expect some variants to be quite common. We recently applied this methodology to show that synonymous mutations in human genes that disrupt exonic splice enhancer motifs are often under strong selection and affect many synonymous sites in our genes.10 This has implications for both diagnostics and for transgene design for gene therapy, as we often remove introns in heterologous genes, so freeing up these residues from their role in specifying exons ceases.11
The same DFE methodology cannot easily be applied to viruses, as the methods assume free recombination (ie, we assume one mutation does not impact the fate of others in the same genome). However, other population genetical tools can still be applied. Recently, we examined SARS-CoV-2 and identified the profile of mutations that we see at four-fold degenerate sites.12 From this profile we could estimate what the synonymous site composition would be, assuming that the only forces are mutational biases and neutral evolution (ie, no selection). We observed that in this genome there is a strikingly strong C->U mutation bias and a G->U one. In the raw data this is not so obvious as G and C are quite rare. However, the mutability of the sites per occurrence of the site reveals the underlying patterns.
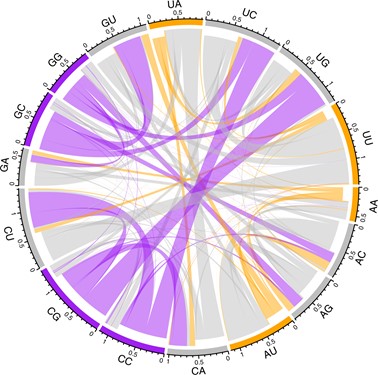
Figure 2: The rate of mutational flux from one dinucleotide to another in the coding sequence of SARS-CoV-2. The direction of flux is indicated by the indentation of the connecting links: the inner layer represents flux out while the outermost layer represents flux into the node. The frequency of the flux exchange is represented by the width of any given link where it meets the outer axis. Dinucleotide nodes are coloured according to their GC-content. Hence, it is evident that there is high flux away from GC-rich dinucleotides whereas AU-rich dinucleotides are largely conserved.12
With knowledge of the mutational bias we then asked what the equilibrium frequency of the four nucleotides would be using four simultaneous equations. This is the nucleotide content at which for every mutation changing a particular base there is an equal and opposite one creating the same base somewhere else in the genome, ensuring overall unchanged nucleotide content. Given the strong C->U and G->U mutational biases, it is no surprise that the equilibrium content is very U rich (we estimate equilibrium U content should be about 65 percent). However, while the four-fold sites are indeed U rich, they are not that U rich, being closer to 50 percent. A clue as to why the mutation bias is so skewed to generating U comes from analysis of equilibrium UU content: UU residues are predicted to be very common, with CU residues being particularly mutable generating UU (Figure 2) – this is expected due to human APOBEC proteins attacking and mutating/editing the virus.13
One probable explanation for this difference between predicted and observed nucleotide content is selection against U content. There may be many U residues appearing in the population, but many are pushed out of the population owing to purification selection, ie, because of the deleterious effects of the mutations. That such selection is happening in the SARS-CoV-2 genome is also clear from the sequence data. We estimate that for every 10 mutations that appear in the sequence databases, another six are lost because of selection prior to genome sequencing. Indeed, UU content is about a quarter of that predicted (Figure 3).
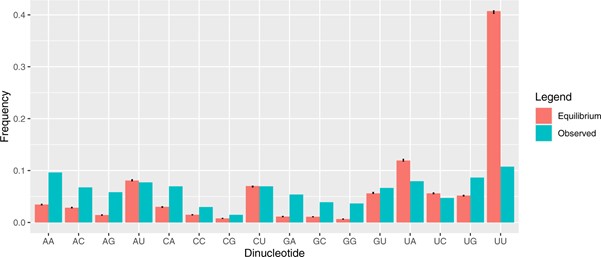
Figure 3: The predicted (under neutral mutational equilibrium) and observed dinucleotide content of SARS-CoV-2. Note the very high predicted levels of UU given the strong mutational flux to UU residues (see Figure 2) and the net underrepresentation in actual sequence.9
This leaves two problems: why is selection operating on SARS-CoV-2 and what can we do with this information? In some cases, we have a good idea as to why: many mutations to U at codon sites generate stop codons. However, we have observed that U destabilises the transcripts and is associated with lower-reported transcript levels;12 a full explanation of the causes of selection on nucleotide content therefore requires manipulation of the sequences.
The second question, what to do with this information, is perhaps more urgent. It has previously been noted that nucleotide content manipulation is a viable means to attenuate viruses.7 Currently there are three groups investigating this route to make a vaccine for SARS-CoV-2: Indian Immunologicals Ltd/Griffith University, Codagenix/Serum Institute of India and Acıbadem Labmed Health Services/Mehmet Ali Aydinlar University. In prior attempts, attention has been paid to CpG levels and UpA levels (which we find to be correlated between SARS genes and between different viruses).12 CpGs attract the attention of zinc antiviral protein (ZAP) and UpA attracts an RNAase L. Not surprisingly, some viruses, including SARS-CoV-2, therefore have low levels of both dinucleotide pairs given the levels of the underlying nucleotides.
The challenge is knowing not just which synonymous sites can be altered but knowing how they should be altered”
In the past, attenuation strategies have focused on modulating synonymous sites to increase CpG and UpA, making the virus more visible to antiviral proteins.14 We in turn suggest a general strategy to utilise this method and to increase U content as well.12 Given the evidence that selection on the virus is to reduce U content, while our antiviral proteins are mutating it to increase U content, it will be interesting to see if we can learn a lesson from nature as to how to weaken a virus. This is an unusual circumstance in which we predict that we should build in more of the already most common synonymous site nucleotides (U in this case) to degrade the virus. More generally, it is assumed that the most used codons are those that tend to increase the fitness of the organism. In the face of such a severe mutation bias, however, this simpler logic no longer holds.
About the author
Laurence D Hurst is Professor of Evolutionary Genetics and Director of the Milner Centre for Evolution at the University of Bath. He is currently also the President of the Genetics Society. He completed his D.Phil in Oxford, after which he won a research fellowship and then moved to Cambridge University as a Royal Society Research Fellow. While on the fellowship he assumed his current Chair at Bath University. In 2015 he was elected a Fellow of the Academy of Medical Sciences and a Fellow of the Royal Society. He is a recipient of the Genetics Society Medal and the Scientific Medal of the Zoological Society of London.
References
- Fath S, Bauer AP, Liss M, Spriestersbach A, Maertens B, Hahn P, et al. Multiparameter RNA and codon optimization: a standardized tool to assess and enhance autologous mammalian gene expression. PLoS One. 2011;6(3):e17596.
- Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22(7):346-53.
- Bentele K, Saffert P, Rauscher R, Ignatova Z, Bluthgen N. Efficient translation initiation dictates codon usage at gene start. Mol Syst Biol. 2013;9:675.
- Kudla G, Murray AW, Tollervey D, Plotkin JB. Coding-Sequence Determinants of Gene Expression in Escherichia coli. Science. 2009;324(5924):255-8.
- Mordstein C, Savisaar R, Young RS, Bazile J, Talmane L, Luft J, et al. Codon Usage and Splicing Jointly Influence mRNA Localization. Cell Syst. 2020;10(4):351-62 e8.
- Stachyra A, Redkiewicz P, Kosson P, Protasiuk A, Gora-Sochacka A, Kudla G, et al. Codon optimization of antigen coding sequences improves the immune potential of DNA vaccines against avian influenza virus H5N1 in mice and chickens. Virol J. 2016;13(1):143.
- Coleman JR, Papamichail D, Skiena S, Futcher B, Wimmer E, Mueller S. Virus attenuation by genome-scale changes in codon pair bias. Science. 2008;320(5884):1784-7.
- Goodman DB, Church GM, Kosuri S. Causes and effects of N-terminal codon bias in bacterial genes. Science. 2013;342(6157):475-9.
- Boel G, Letso R, Neely H, Price WN, Wong KH, Su M, et al. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature. 2016;529(7586):358-63.
- Savisaar R, Hurst LD. Exonic splice regulation imposes strong selection at synonymous sites. Genome Res. 2018;28(10):1442-54.
- Thumann G, Harmening N, Prat-Souteyrand C, Marie C, Pastor M, Sebe A, et al. Engineering of PEDF-Expressing Primary Pigment Epithelial Cells by the SB Transposon System Delivered by pFAR4 Plasmids. Mol Ther Nucleic Acids. 2017;6:302-14.
- Rice AM, Castillo Morales A, Ho AT, Mordstein C, Mühlhausen S, Watson S, et al. Evidence for strong mutation bias towards, and selection against, U content in SARS-CoV-2: implications for vaccine design. Molecular Biology and Evolution. 2020:(in press).
- Simmonds P. Rampant C->U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses – causes and consequences for their short and long evolutionary trajectories. bioRxiv. 2020:2020.05.01.072330.
- Tulloch F, Atkinson NJ, Evans DJ, Ryan MD, Simmonds P. RNA virus attenuation by codon pair deoptimisation is an artefact of increases in CpG/UpA dinucleotide frequencies. Elife. 2014;3:e04531.
Related topics
Disease Research, DNA, Gene Therapy, Genetic Analysis, Genomics, Protein, Proteogenomics, Proteomics, Research & Development, RNAs, Vaccine
Related conditions
Covid-19