New target for regulating mitochondria during stress identified
Posted: 19 September 2019 | Rachael Harper (Drug Target Review) | No comments yet
A new target has been identified for the treatment of heart failure, heart attack, stroke and neurodegeneration.
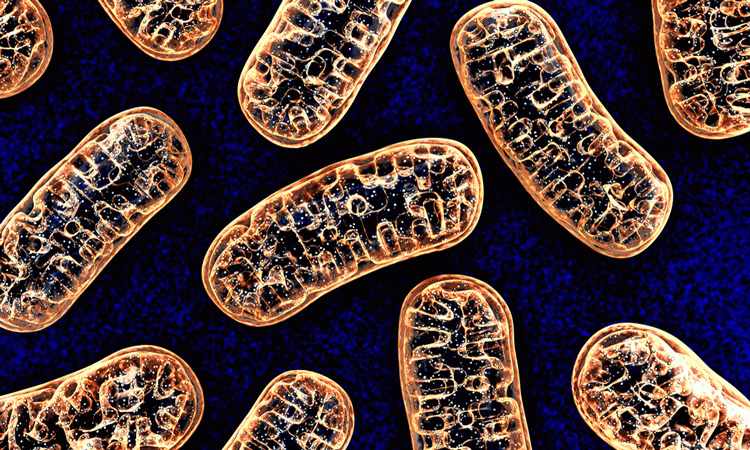
Researchers from the Lewis Katz School of Medicine at Temple University, US have shown for the first time that a specialised emergency responder protein (which are activated in the heart during a heart attack to help prevent cell death), known as MCUB, temporarily decreases harmful levels of calcium transport into mitochondria, the energy-generating batteries of cells.
This identifies MCUB as a promising new target for the investigation and treatment of conditions that feature calcium overload and cell death – conditions that include heart failure, heart attack, stroke and neurodegeneration.
“MCUB fine-tunes calcium uptake by mitochondria in injured heart tissue, in an attempt to limit calcium overload, which is a major contributor to cell death, particularly following a heart attack,” explained John W Elrod, PhD, Associate Professor in the Center for Translational Medicine at the Temple University Lewis Katz School of Medicine and senior investigator on the new study.
Dr Elrod’s team found that deletion of the MCUB gene in cells results in a change in the proteins that are essential for controlling whether the channel is on or off. Since these alterations are induced by stress, the researchers next investigated the role of MCUB after heart attack in mice.
They observed significant elevations in MCUB gene expression and decreases in MCU and the gatekeeper of the channel, MICU1. When genetically expressed prior to inducing a heart attack in mice, MCUB altered the channel to reduce calcium overload in the injured heart, ultimately curtailing tissue injury.
“MCUB presents us with a new molecular target for investigation,” Dr Elrod said. “It’s unique in that it alters the stoichiometry of the channel and thereby presents a new mechanism which may be amendable to therapeutic manipulation. We think that modulating MCUB may allow us to tune down mitochondrial calcium uptake without completely inhibiting all energetic function.”
It is hoped that follow-up studies defining the exact sites of molecular interaction will provide additional insight into how to target mitochondrial calcium overload in heart disease.
The research was published in the journal Circulation.
Related topics
Drug Targets, Molecular Biology, Protein, Research & Development
Related conditions
Heart attack
Related organisations
Lewis Katz School of Medicine at Temple University
Related people
John W Elrod PhD